Last week the CHI Drug Discovery
Chemistry (DDC) meeting returned triumphantly to San Diego. This was the best
conference I’ve attended in years, which reflects not just the quality of the
meeting itself but the fact that three-dimensional events are vastly superior
to their 2D counterparts.
About 75% of the more than 700
attendees were physically present, though having a virtual option turned out to
be wise; at least three of the speakers had COVID but were still able to
present remotely. Although the FBDD track lasted just a day and a half,
fragments were well-represented across the four days and ten tracks. I won’t
attempt to summarize the more than 40 talks I attended but will just cover some
broad themes.
Computational Methods
Seva Katritch (USC) described the
V-SYNTHES approach we highlighted in January. This modular method enables
computational fragment growing, in effect facilitating a search of 11 billion
molecules from just 600,000 scaffolds. The method as described makes heavy use
of Enamine’s make-on-demand molecules, and I think everyone in the audience was
excited to hear that the company has started making and shipping compounds from
Kyiv again.
In the comments to the blog post
on V-SYNTHESES someone mentioned BioSolveIT, and Paul Beroza (Genentech)
described using their software for a similar approach. One of the targets they investigated,
ROCK1, was also investigated with V-SYNTHES, and both techniques yielded unique
nanomolar inhibitors.
Jan Wollenhaupt (Helmholtz
Zentrum Berlin) also mentioned BioSolveIT in the context of fragment growing by
catalog. Fragments identified crystallographically from their F2X libraries (see
here) were grown to low micromolar endothiapepsin binders. Interestingly an
unbiased docking screen did not find these molecules, illustrating the utility
of stepwise computational approaches.
DOTS is another approach to
computational growing and docking enabled by rapid synthesis we’ve previously
written about, and Xavier Morelli (CNRS) gave an update, including the fact
that they plan to launch a webserver soon.
Physical Methods
Tim Kaminski (InSingulo)
described an intriguing method for screening liposome-bound proteins such as
GPCRs. Dyes incorporated into the liposome are visualized using single molecule
microscopy, and the liposomes can be observed in real time binding to
immobilized targets in 384-well plates. Tim mentioned that the instrument
should be available for purchase next year.
A new take on an old method was
described by Félix Torres (ETH), who discussed using photochemically induced
dynamic nuclear polarization (photo-CIDNP) to increase the sensitivity of NMR,
thereby reducing experimental times by a factor of 100. The method requires
specialized fragments and a customized NMR, but they can currently screen 1500
fragments per day, and the approach could be particularly valuable for
screening hard-to-express proteins.
Sticking with the theme of photochemistry,
Rod Hubbard (Vernalis by way of Hitgen) discussed a DNA-encoded library of more
than 130,000 fragment-linker combinations each containing a photoaffinity tag.
Screening this against PAK4 yielded 425 hits, and of the 30 chosen for
validation more than 90% confirmed by NMR or crystallography. As we noted in
2020, combining DEL and FBLD provides new opportunities for exploring chemical
space.
It’s been a few years since we
discussed weak affinity chromatography, and Kirill Popov (WAC) provided an
update. They’ve applied the approach to more than 50 targets and have obtained hit
rates up to 20%. An example against SMARCA4 yielded hits that were subsequently
found to bind at two sites, one of which had not previously been described.
Covalent fragments continue to
increase in popularity. FragNet alum Lena Muenzker (BI) described an intact-protein
mass spectrometry screen of the E3 ligase SIAH1 against 1260 acrylamides,
resulting in 214 hits. Crystallography has been successful, and they are planning
to use these to generate covalent PROTACs.
We’ve previously written about screening
covalent fragments in cells, and Benjamin Horning (Vividion) and Madeline
Kavanagh (Scripps) described a nice chemoproteomics case study in which an
alkynamide-containing fragment was identified that binds to cysteine 817 in the
kinase JAK1. Optimization led to a low nanomolar binder that inhibits JAK1
signaling.
Cysteine is not the only amino
acid amenable to covalent modification. Plenary keynote speaker Laura Kiessling
(MIT) described squarate derivatives as tunable “Goldilocks” warheads for
lysine, with the right balance of reactivity and stability.
Success Stories
Cases studies were abundant,
including some new disclosures that I’ll hold off describing until they publish.
Of course, drugs are the ultimate success stories, and several of these were presented.
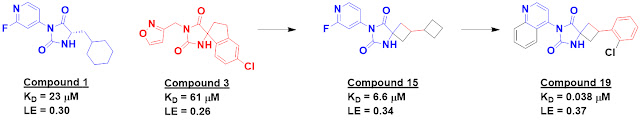
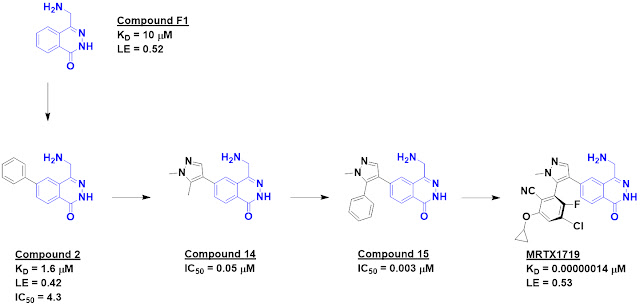
Svitlana Kulyk recounted the discovery of MRTX1719, Mirati’s MTA-cooperative
PRMT5 inhibitor, including some interesting tangents not discussed in the
publication.
Steve Fesik (Vanderbilt) gave two
presentations on near-clinical compounds, one targeting MCL1 and the other WDR5.
In both cases weak fragments were advanced to picomolar binders within one to
two years, but it has taken much longer to optimize other properties of the
molecules.
Indeed, this turned out to be something
of a theme. Valerio Berdini (Astex) discussed the discovery of erdafitinib, the
third approved FBLD-derived drug. The program started in 2006, and it took just
nine months to go from the fragment hit to late lead optimization. But the compound
didn’t enter the clinic until 2012, and it took until 2019 to be approved.
Similarly, Wolfgang Jahnke
(Novartis) described the story of the sixth approved fragment-based drug. The
fragment screen against ABL was conducted in 2006, but the project went through
two near-death experiences. Asciminib finally entered the clinic in 2014, and
it was approved last year.
But timelines are not destined to
be long. We’ve previously written about vemurafenib, the first FBLD-derived
drug, which took just six years from project initiation to approval. Ryan Wurz
(Amgen) gave a retrospective on sotorasib, the fifth approved FBLD-derived
drug. Amgen started the program in August 2012, sotorasib was first synthesized
in early 2017, first dosed in humans in 2018, and approved in May of last year.
Fast doesn’t mean easy: it took 110 co-crystal structures, and I counted more
than 100 names on the acknowledgement slide. But success against KRAS is a
welcome reminder that sometimes we really can accomplish the impossible when we
work together.
This is a good point on which to
close. Assuming SARS-CoV-2 doesn’t intervene, DDC is scheduled to return to San
Diego April 10-13 next year. I hope to see you there!