Today marks exactly two years
since Practical Fragments first mentioned SARS-CoV-2. Since then,
COVID-19 has killed more than 6 million people worldwide. Multiple effective
vaccines have been developed and approved, along with a couple small-molecule
drugs, but the virus is here to stay, and more drugs will be needed. This
brings us to an open-access paper published in J. Am. Chem. Soc. by Jens
Carlsson (Uppsala University) and a large group of international collaborators.
The so-called main protease (Mpro,
or 3CLp) has been an antiviral target since the earliest days of the pandemic;
the work we highlighted two years ago focused on a crystallographic screen
against this enzyme. The new paper describes two virtual screening approaches.
The first started with a library
of 235 million virtual compounds, mostly from Enamine’s “readily available for
synthesis” (REAL) collection. Each compound was docked in thousands of
different orientations against the active site of Mpro using
DOCK3.7. Despite the staggering numbers (more than 223 trillion complexes!), the
screen took just a day on 3500 CPU cores. The top 300,000 compounds were clustered
based on similarity, and 100 molecules were synthesized. Nineteen of these
showed binding by SPR, and three also inhibited the enzyme. Crystal structures
were obtained for two of these, and both bound similarly to the predicted
binding modes.
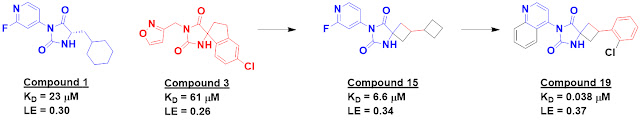
Compounds 1 and 3 each contain a
hydantoin moiety that makes multiple hydrogen bonds to the protein, and merging
elements led to low micromolar compounds such as compound 15. Further optimization
ultimately delivered compound 19.
Compound 19 was potent in SPR and
biochemical assays. Though it binds noncovalently, it had comparable cellular activity to nirmatrelvir, the recently approved covalent inhibitor of Mpro.
Compound 19 showed nanomolar cell potency against SARS-CoV-1 and MERS-CoV
and good selectivity against ten human proteases. The in vitro stability and
permeability of compound 19 are also promising.
In addition to this de novo
virtual screen, the researchers performed a second screen starting from one of
the fragments identified crystallographically at Diamond Light Source. Of 93
molecules purchased and experimentally tested, 21 showed binding by SPR and 5
of these also inhibited the enzyme, with the most potent compound showing low
micromolar activity.
There are several lessons from this
paper. First, despite searching hundreds of millions of compounds, the best
hits had only modest activity. This is perhaps surprising given the high fragment
hit rates observed against Mpro in crystallographic and NMR screens,
though it is worth noting that those fragments were even weaker binders.
Second, the hit rate from the naïve
virtual screen was similar to that from the experimentally derived fragment
screen. The researchers suggest that perhaps docking “may be more proficient in
ranking diverse chemotypes rather than differentiating between closely related
elaborations of the same scaffold.” In other words, virtual screens seem better
at evaluating diverse starting points rather many similar molecules.
Third, despite the fact that the de
novo virtual screen was not explicitly fragment-based, compound 1 does actually
adhere to the rule of three. From there, addition of just six atoms improved
affinity by >600-fold while also improving ligand efficiency.
Finally, this work is a testament
to the utility of combining massive virtual screening with readily
synthesizable compounds: the researchers note that it took less than four months
to progress from compound 1 to nanomolar inhibitors.
This work relied heavily on rapid
chemical synthesis done in Ukraine. Indeed, the two most popular fragment
suppliers are both largely based in that country. Over the years many of us have
come to know Ukrainian scientists not just as trusted colleagues but also as
friends. I wish them and their families safety, and strength.


No comments:
Post a Comment