The biotech industry rightly
prizes speed: every day people die of diseases we are trying to prevent or
cure. And developments can indeed happen quickly. Just eight years elapsed from
the demonstration that a mutant form of KRAS was druggable to the approval of
sotorasib, with less than three of those years spent in the clinic. Even more dramatically,
it took less than a year from the first reports of SARS-CoV-2 to develop
effective vaccines. But as two recent pieces in Nature Rev. Drug Disc.
demonstrate, such speed is not necessarily the norm.
The first, by Asher Mullard, is entitled
“FDA approves 100th monoclonal antibody product.” This is a nice
review of a remarkably successful therapeutic approach. But this triumph was not
a foregone conclusion. Mullard traces the field’s origin to the mid-1970s, and
while the first drug was approved in 1986, it took another eight years for the
second. The article includes a timeline showing approvals by year, and it is interesting
to compare this with FBDD-derived drug approvals since the 1996 publication of the
seminal SAR by NMR paper. In the chart below, the first year on the x-axis is for antibody drugs; the second is for FBDD-derived drugs.
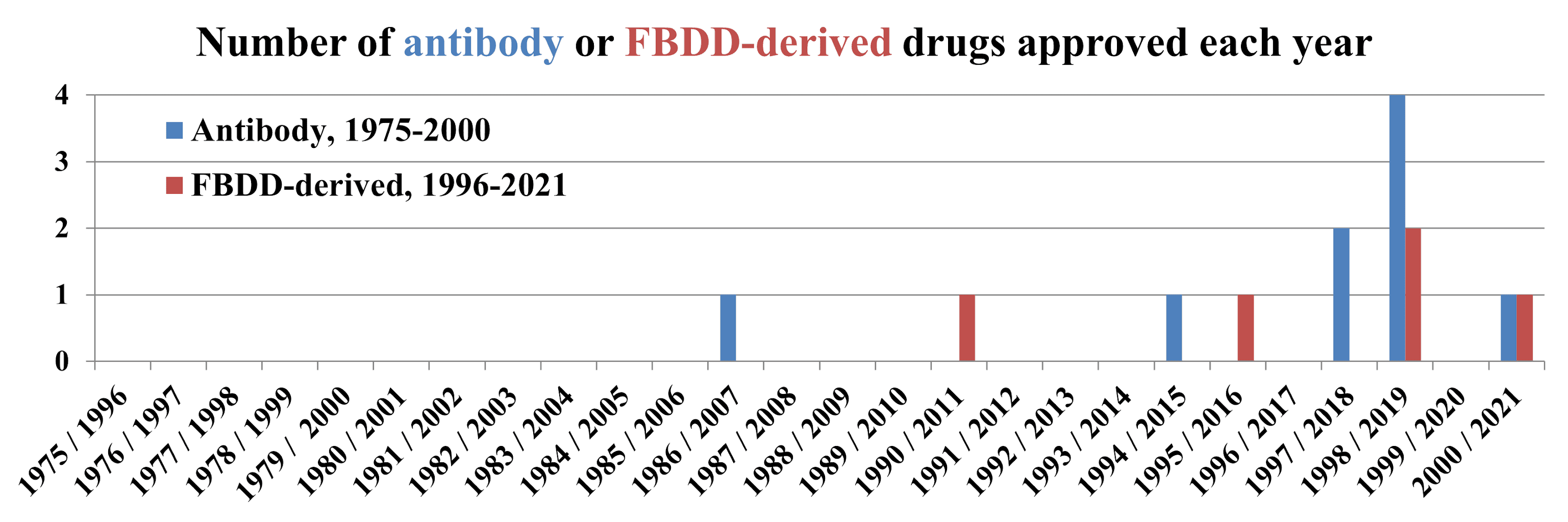
A quarter century after work
began, new antibody approvals were still uncommon; Mullard notes that “antibody
approvals have only been an annual event since 2006.”
Antibody-drug conjugates (ADCs)
are an interesting subset that – as their name suggests – comprise an antibody
linked to a small molecule, usually a toxin intended to kill cancer cells. Ten of
these have been approved in the US, but while the first (gemtuzumab ozogamicin)
was approved in 2000 most of the rest are recent, with six of them coming since
the beginning of 2019.
By these standards the fact that
only five fragment-derived drugs have been approved thus far isn’t surprising.
Indeed, antibodies have some advantages: “whereas medicinal chemists
can toil for years to find small molecules with activity against a given
target, antibody discovery can take a matter of months.” Moreover, as the
article continues, success in the clinic is roughly double that of small
molecules.
The second article is by
Christopher Austin, until recently Director of the National Center for
Advancing Translational Sciences at the US National Institutes of Health. Titled
“Translational Misconceptions,” it briefly enumerates and debunks false beliefs
about translating new discoveries into drugs, which include:
- Translation does not exist
- Translation is a “thermodynamically favored” process
- Translation is straightforward and does not qualify as science
- Translation is a unidirectional process
- Once an investigational therapy gets into humans for the first time, regulatory approval
and marketing are all but assured
- Regulatory approval is the end of the translational process
Those of us in industry would probably
dismiss these statements as naïve, but such perceptions are widespread. Indeed,
Austin himself acknowledges that he “once believed unquestioningly in all of
them.”
Each of these misconceptions
invites discussion. To take just the last, the first approved ADC was pulled
from the US market in 2010 when confirmatory trials showed that patients on the
drug actually did worse than those on placebo. It was reapproved in 2017 after
a better dosing schedule was established. In other words, it took 17 years
after initial approval to figure out how to effectively use gemtuzumab
ozogamicin, and 26 years from the beginning of the project.
Returning to the two successes mentioned at the top of this post reveals that their apparent rapidity does not tell the full story. The Tethering technology that eventually
led to sotorasib was initially published more than twenty years ago, and researchers
first used mRNA packaged in liposomes to transfect cells way back in 1989.
Amidst rapid visible progress it
is easy to lose sight of the fact that much research goes nowhere very
slowly. Even when successful, it might take decades to help patients. As Austin concludes, “only by advancing our common understanding of the
complexity of translation, translational research and translational science
will translational gaps be narrowed and eventually eliminated.”





