Bromodomains, which recognize acetylated
lysine residues, are popular cancer targets due to their role in gene
regulation. A plethora of potent inhibitors have been reported, many of them
derived from fragments, and some have even gone into the clinic. The story
behind one of these, ABBV-744, was recently published in Nature by Yu
Shen and colleagues at AbbVie.
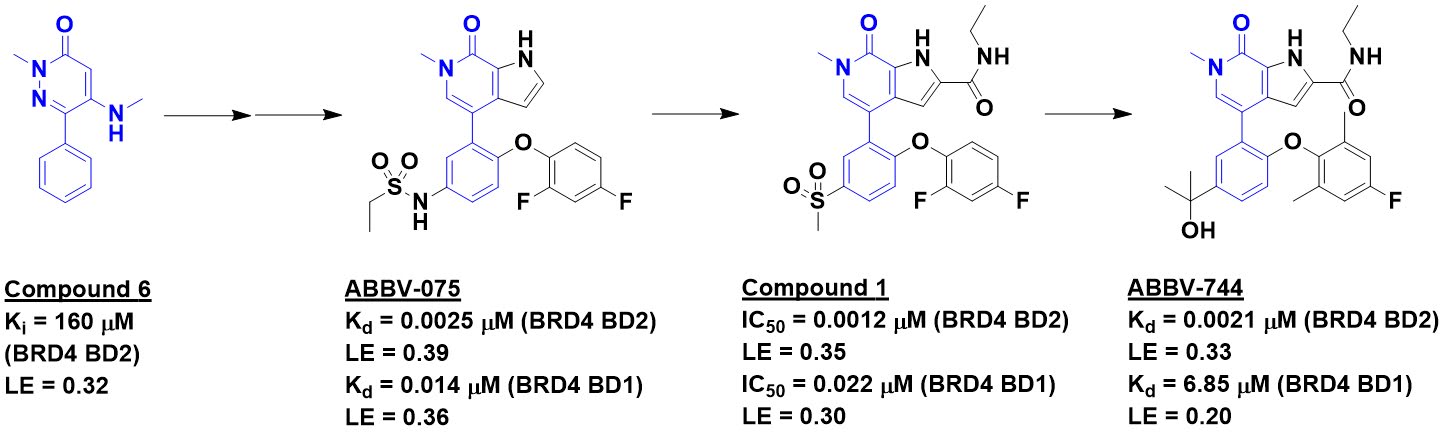
The story starts with a protein-detected
NMR screen (highlighted here), which ultimately led to ABBV-075 (highlighted here).
This molecule binds tightly to the four BET-domain family members (BRD1, BRD3, BRD4,
and BRDt). However, ABBV-075 causes gastrointestinal toxicity as well as a
reduction in platelets when tested in mice. Indeed, these effects are seen when
BRD4 alone is genetically silenced in mice, suggesting on-target toxicity. However,
each of the BET proteins has two separate bromodomains, called BD1 and BD2, and
the researchers thought that a selective inhibitor of the BD2 domain might be
better tolerated.
Screening about 2500 compounds
from the ABBV-075 program revealed that compound 1 was still quite potent
against the BD1 domain of BRD4 but lost activity against the BD2 domain.
Further optimization ultimately led to ABBV-744, which is at least two orders of magnitude more
selective against the BD2 domains of all four BET-domain proteins over the
respective BD1 domains. It also shows no activity against a panel of kinases and
other bromodomains, and is orally bioavailable. A crystal structure of the
molecule bound to either BD1 or BD2 reveals that the key interactions seen in ABBV-075
are maintained, but that the added amide moiety makes interactions only
available in BD2, while the larger diphenylmethyl ether moiety is better accommodated
in BD2 due to a slightly larger pocket (containing a valine rather than an
isolueucine residue).
ABBV-744 is active against multiple acute myeloid leukemia and prostate cancer cell lines, and the paper thoroughly explores the biology of a selective BD2 inhibitor. Most striking is that in a mouse xenograft model, ABBV-744 shows similar activity at 1/16 of its maximum tolerated dose (MTD) as ABBV-075 shows at its MTD. Even at doses well above efficacious exposure levels, ABBV-744 shows only limited platelet reduction and no gastrointestinal toxicity in mice. As mentioned at FBLD 2018, this molecule has entered clinical development, while ABBV-075 has quietly been dropped from AbbVie’s pipeline.
This is a lovely example of
biology-guided medicinal chemistry that is reminiscent of the BCL-family
inhibitors, which started with less specific molecules and culminated with the
approval of BCL2-selective venetoclax. Although the fragment origins of ABBV-744
are clear, they are not mentioned in the paper and – like the KRAS inhibitors
and AZD5991 – could be easily overlooked. In all these cases small starting
points have delivered potentially huge drugs, and Practical Fragments
wishes everyone involved the best of luck.