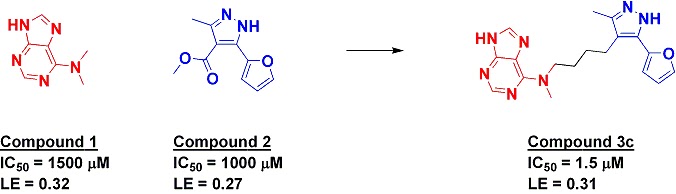
Linking two weak fragments to get
a potent binder is something many of us hope for. Unfortunately, as a poll
taken a few years back indicates, it often doesn’t work. But why? This is the
question tackled by Lingle Wang and collaborators at Schrödinger and D. E. Shaw
in a recent J. Chem. Theory Comput. paper.
When a ligand binds to a protein
it pays a thermodynamic cost in terms of lost translational and orientational
entropy. By linking two fragments, this cost is paid only once instead of
twice. In theory this should lead to an improvement of 3.5-4.8 kcal/mol in
binding energy, resulting in a 400-3000-fold improvement in affinity over what
would be expected from simple additivity. As we noted here, this is possible,
though rare. Linker strain often takes the blame as a primary villain. But is there
more to the story?
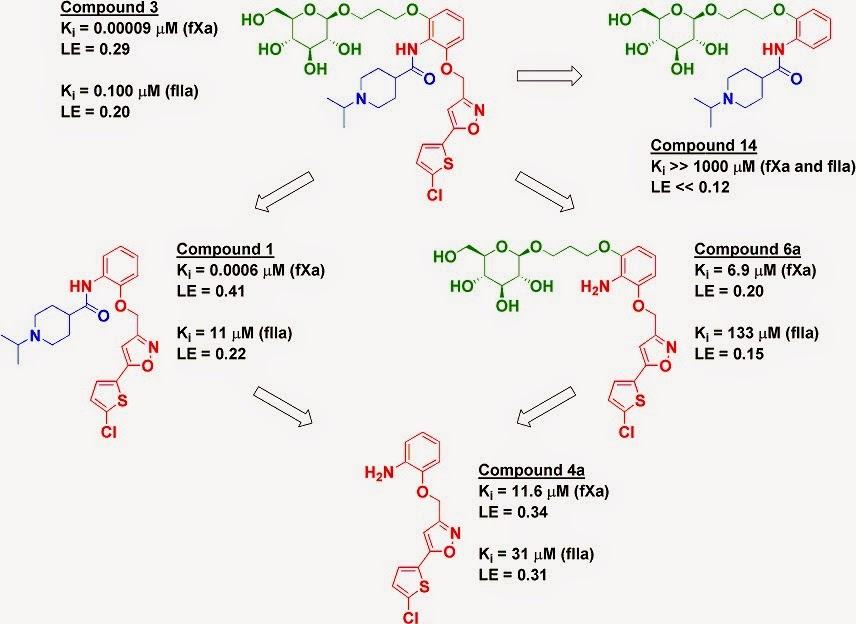
The researchers computationally
examined published examples of fragment linking (most of which we’ve covered on
Practical Fragments) using free energy perturbation (FEP) to try to
understand why the linked molecules bound more or less tightly than expected.
Impressively, they were able to computationally reproduce experimentally
derived numbers, and by building a thermodynamic cycle they could extract the
various components of the “connection Gibbs free energy.” These included
changes in binding mode or tautomerization, linker strain or linker interactions
with the protein, and the previously mentioned entropic benefits of fragment
linking.
The analysis also identified two
additional components. If two fragments favorably interact with each other,
covalently linking them may not give as much of a boost. This concept had been considered decades ago, though the current work provides a more general understanding.
The more important factor appears
to be what the researchers refer to as “configurational entropy.” The notion is
that even when a fragment is bound to a protein, both the ligand and protein
retain considerable flexibility, which is entropically favorable. Linking two
fragments reduces the configurational entropy of each component fragment, and
the linked molecule binds less tightly than would be expected. The researchers
argue that this previously unrecognized “unfavorable change in the relative
configurational entropy of two fragments in the protein pocket upon linkage is
the primary reason most fragment linking strategies fail.” They advise that
maintaining a bit of flexibility in the linker can help, as has been previously suggested.
This is an interesting analysis,
and explicitly considering configurational entropy is likely to improve our
understanding of molecular interactions. But is it really the main barrier to
successful fragment linking? The researchers explore only nine different
protein-ligand systems, though they did consider multiple linked molecules for
three of these (pantothenate synthetase, RPA, and LDHA). Still, these represent
just a fraction of the 45 examples collected in a recent review, and they only
considered one somewhat contrived case (avidin) in which especially strong
superadditivity was observed. It will be interesting to see whether the
analysis holds true for more examples of fragment linking.