Last week we highlighted three chemistry-focused
papers, and this week we’ve got three more. And to add a third three, all these
papers address “three-dimensional” fragments.
The first, in ACS Med. Chem.
Lett. by Brian Cox (University of Sussex), Philip Cox (AbbVie) and
colleagues, describes using photochemical [2 + 2] cycloadditions to generate
bridged pyrrolidine fragments, which can be further diversified.

The researchers analyzed 54
products by principal moments of inertia (PMI) and plane of best fit (PBF),
which revealed them to be quite shapely, more so than AbbVie’s Rule of Three
collection. Interestingly, amide derivatives with aromatic substituents were
often shapelier despite being less saturated, and the researchers thus caution
against using Fsp3 as a proxy for shapeliness – a point Teddy made
several years ago.
The next paper, open access in ChemComm,
also deals with cyclobutane-containing molecules. However, rather than building
them from scratch in cycloaddition reactions, David Spring and collaborators at
University of Cambridge and California State Polytechnic University Pomona use
palladium-catalyzed C-H arylation to functionalize the rings. Lactonization and
further derivatization generates a range of molecules.

The researchers generated a
virtual library of 90 scaffolds that were rule-of-three compliant and quite
shapely. It would be interesting to explore this chemistry on the bridged pyrrolidines
of the previous paper – perhaps deliberately “losing control,” as discussed
last week.
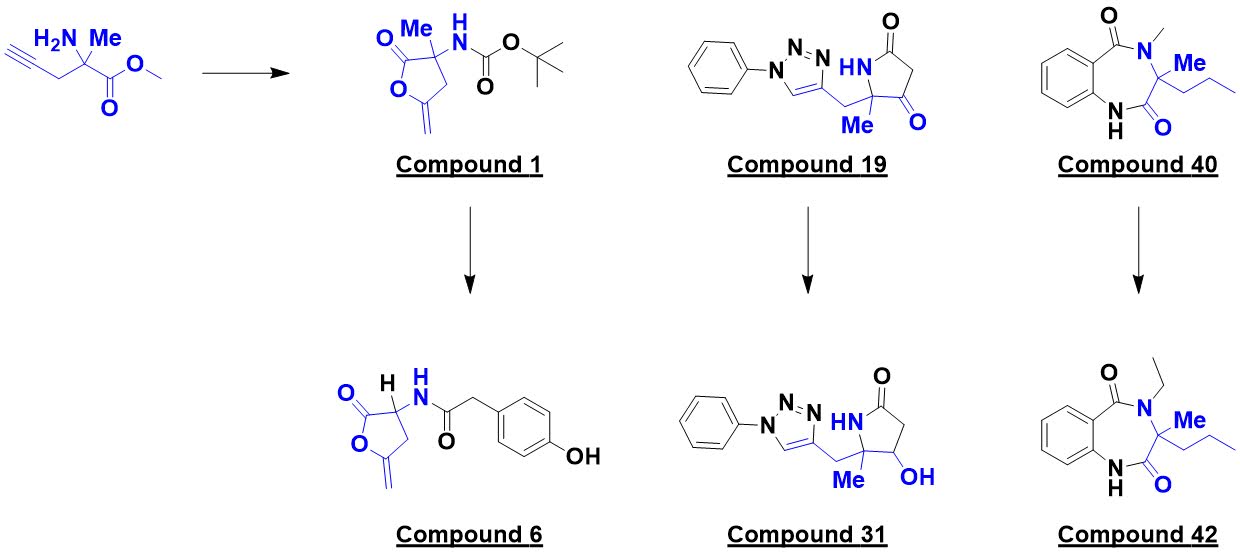
Finally, in Chem. Eur. J.,
Peter O’Brien (University of York) and a large group of collaborators from academia
and industry describe (also open access) the “design and synthesis of 56 shape diverse 3-D
fragments.” Because of their prevalence in drugs, pyrrolidines and piperidines
were chosen as targets. The researchers specifically set out to make diverse
molecules that would be shapelier (as assessed by PMI) than members of typical
libraries. In considering three-dimensionality, the researchers considered not
just the lowest energy conformation, as is typically done, but also other conformations
with energies up to 1.5 kcal/mol higher; these would be present roughly 8% of
the time at 37 °C. Some of the molecules are shown.

A PMI analysis of these fragments
revealed them to be more three-dimensional than representatives of six
commercial fragment libraries. In fact, although three of the commercial libraries
are touted as being “3D,” a PMI analysis revealed them to “have only a
marginally better 3-D profile compared to the standard 2-D rich commercial
fragment libraries.” As in the paper discussed above, there was no correlation
between Fsp3 and PMI.
Despite being relatively simple,
42 of these molecules had been previously unreported. In addition to their shapeliness,
they also adhered to the rule of three, with an average ClogP of just 0.54.
Moreover, 52 of the fragments were stable for >6 weeks in DMSO, 48 were
stable in aqueous buffer for >24 hr, and 40 of them were soluble at >0.5 mM in buffer.
Most of these fragments are
available for screening at the Diamond XChem facility. It will be interesting
to see what kinds of hit rates they produce, and whether they generate
superior leads. As we noted last year, the majority of drugs are not particularly
shapely. Still, it is fun to explore new regions of chemical space, and these
three papers are good starting points.