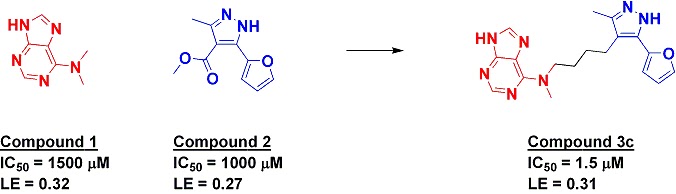
The researchers start by briefly reviewing nine published examples of fragment linking where affinities for both fragments as well the linked molecule are provided (some of these have been discussed previously here, here, and here). Of these, only three examples showed clear superadditivity (in which the linked molecule has a significantly higher affinity than the product of the affinities of the individual fragments), and two of these examples are rigged systems in which a molecule already known for its potency (such as biotin) is dissected into fragments. The challenges of linking are succinctly summarized:
The keys to achieving superadditivity upon linking are to maintain the binding modes of the parent fragments, not introduce both entropy and solvation penalties while designing the linker, and also make any interactions with the intervening protein surface that need to be made.Also, of course, the resulting molecule needs to be synthetically accessible. Having a certain amount of flexibility in the linker can be useful, as this will allow the fragments some room to shift around, but too much flexibility introduces an entropic cost that defeats the purpose of linking in the first place. Software tools such as those by BioSolveIT can help design the linker, but what if some fragments themselves are inherently better suited for linking?
All three of the examples that show superadditivity start with one fragment that is highly polar and makes hydrogen bonds or metal-mediated bonds with the protein. The researchers suggest that such fragments are likely to pay a heavy thermodynamic penalty when they are desolvated, and that this cost can be reduced by linking them to a hydrophobic fragment. Thus, to maximize your chances of successful linking, the authors suggest you should choose
a fragment pair that consists of one fragment that binds by strong H-bonds (or non-classical equivalents) and a second fragment that is more tolerant of changes in binding mode (hydrophobic or vdW binders).
This is an interesting proposal, though because there are so few examples it is hard to assess. Indeed, the only other case of clear superadditivity I found involves dimerizing a fragment that is reasonably hydrophobic (ClogP = 2.4), albeit negatively charged. Hopefully we’ll see more examples in the coming years, but in the meantime, linking a water-loving fragment to an oily one is worth a shot.