Although 2020 and 2021 were baleful
exceptions, tuberculosis is normally the world’s deadliest infectious disease. The
pathogen Mycobacterium tuberculosis (Mtb) makes its home inside
macrophages, the very cells that normally destroy microorganisms. Worse, some strains
have become resistant to approved drugs. In a recent open-access J. Med. Chem.
paper, Madeline Kavanagh, Kirsty McLean, and collaborators at University of
Manchester, University of Cambridge, and elsewhere explore a new mechanism to
fight this ancient disease.
An important nutrient source Mtb
exploits inside human cells is cholesterol, which bacteria oxidize with the cytochrome
P450 enzyme CYP125. A second enzyme, CYP142, is also present in some strains
and is functionally redundant. Thus, the researchers set out to make a dual
inhibitor.
Mtb has some 20 CYPs, and
the Cambridge researchers have been studying them for a long time: we wrote
about their work on CYP121 in 2016 and their work on CYP126 in 2014. All these enzymes
contain a heme cofactor, and much is known about targeting the bound iron.
However, some ligands are promiscuous, hitting human P450 enzymes, or they are
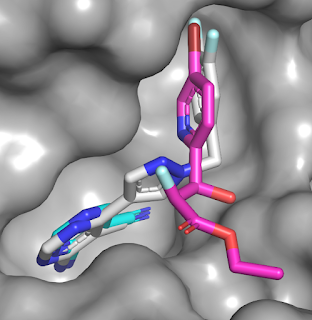
rapidly effluxed out of cells. Thus, the researchers built a fragment library
of just 80 likely heme binders but excluded particularly promiscuous moieties,
such as imidazoles. The library was screened using UV-vis spectroscopy; ligands
that bind to the heme group cause a red-shift in the λmax. Only four
hits were found for CYP125, while a dozen were found for CYP142, including
three of the four CYP125 hits. Compound 1a had modest affinity for CYP125 and
low micromolar affinity for CYP142.

Functional assays revealed that
compound 5m inhibited both enzymes with nanomolar activity, comparable to their
affinities. It also inhibited the growth of Mtb grown on media
containing cholesterol as the sole source of carbon. More impressively, it even
inhibited the growth of Mtb in standard media spiked with just low
concentrations of cholesterol. Oddly though, it also inhibited the growth of Mtb
grown on media not containing cholesterol, albeit at a higher concentration,
suggesting perhaps other targets. But one reason tuberculosis is so hard to
treat is that the bacteria persist inside human cells. Encouragingly, compound 5m
inhibited the growth of Mtb in human macrophages at low micromolar
concentrations, and it did not show
cytotoxicity up to 50 micromolar concentration.
Unfortunately, compound 5m did
show cytotoxicity to human HepG2 cells, and it also inhibited several human P450
enzymes at high nanomolar concentrations, which could cause drug-drug
interactions. Also, selectivity against other MTb P450 enzymes is unclear.
Finally, no in vitro ADME data are reported. Nonetheless, this is a nice
fragment to lead story, and compound 5m could be used – cautiously – as a
chemical probe to study Mtb biology.







.png)



