Two hot areas of FBLD include
covalent fragments (see here for example) and high-throughput crystallography (see
here and here). Two new papers add to the excitement but also reveal some of
the challenges.
The first, published open access in
J. Med. Chem. by Charles Eyermann (University of Cape Town), Christopher
Schofield (Ineos Oxford Institute of Antimicrobial Research), Christopher Dowson
(University of Warwick) and collaborators at Diamond Light Source focuses on an
antibacterial target, the penicillin binding protein PaPBP3 from Pseudomonas
aeruginosa.
Some members of the group had
previously screened a library of 40 compounds against this protein and
identified a single hit that bound covalently. Inspired by this result, they
assembled a library of 262 commercial covalent fragments, 152 of which
contained boron, an element known to react with serine and threonine hydrolases. These were
screened crystallographically at 250 mM, resulting in 34 boron-containing hits “with
various levels of electron density observed at the active site.” Many of these
seem to have given ambiguous density, and only a handful of fragment structures
are reported.
Next, the researchers attempted fragment
growing; the team ultimately obtained 10 structures, drawing from the original
fragments and the elaborated molecules. Interestingly, these showed three
different binding modes: a monocovalent complex with the active-site serine, a
dicovalent complex with the active-site serine and a neighboring serine, and a
tricovalent complex with these two serine residues and a nearby lysine side
chain.
Despite having up to three
covalent bonds to the enzyme, even the elaborated molecules showed at best
double-digit micromolar inhibition of PaPBP3, and none showed antimicrobial
activity. The bond between boron and serine or lysine is reversible, so while
disappointing, this weak activity is not entirely surprising. The result is
reminiscent of efforts against the SARS-CoV-2 main protease, Mpro or
3CLpro, where many of the crystallographic hits turned out to be very weak binders.
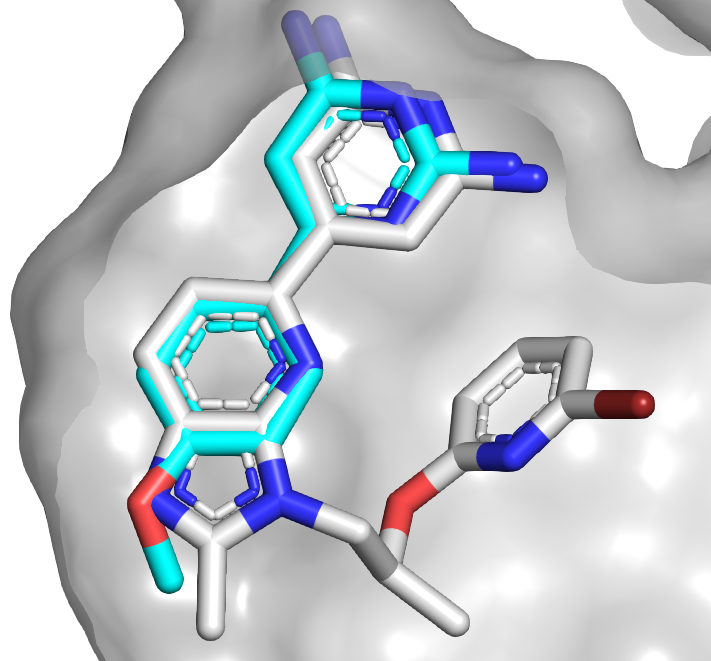
The second paper, published open
access in Angew. Chem. Int. Ed. by Alexander Dömling and collaborators
at University of Groningen and the Paul Scherrer Institute, also looked at 3CLpro.
Here though, rather than starting with commercial fragment libraries the
researchers used high-throughput synthesis to make their own.
The Dömling lab has had a
long-standing interest in multi-component reactions in which several reagents
are combined to generate products. The researchers explored the Passerini reaction (which uses an aldehyde or ketone, a carboxylic acid, and an
isocyanide) and the Ugi reaction (which uses all of these as well as an amine).
For the carboxylic acid, they chose an electrophile such as acrylic acid. They
initially worked out the conditions at 0.5 mmol scale in 96-well plates and
then purified the products using chromatography or precipitation. The majority
of wells yielded product in good yields and purity.
Next, the researchers turned to
high-throughput methods, performing the reactions in 384-well plates using acoustic
liquid handling. This is similar to the approach I wrote about here that was
used to discover covalent KRAS inhibitors, ultimately leading to the approved drug
sotorasib.
The researchers then soaked 181
of their compounds against the SARS-CoV-2 protein 3CLpro, with each compound at
10 mM. Unlike the crude reaction screening described here, the researchers used pure
compounds. This effort resulted in five hits, though one of them
turned out to bind noncovalently. Three of the molecules had low micromolar
activity.
In the end, while both of the
papers do report the discovery of covalent modifiers, the functional activities
in the first paper are modest, and the active warheads in the second paper (chloroacetamides
and acrylates) are likely too reactive to be advanceable. A nice feature of
crystallography is that it can provide structural information for extremely weak
hits. But as we’ve asked previously, how weak is too weak? Getting more crystal structures more rapidly is one thing, but figuring out which ones are
useful requires even more skill, creativity, and luck.