Synthetic lethality is a
relatively new approach to cancer therapy. The idea is to inhibit a protein
that is necessary for cancer cells but dispensable for normal cells, thereby
minimizing toxicity. Last year we described one example, and in a just-published open access
J. Med. Chem. paper Chris Smith and colleagues at Mirati describe
another.
The biology gets a bit
complicated, so please bear with me. Protein arginine methyl transferase 5
(PRMT5) is an epigenetic writer that adds two methyl groups to arginine
residues in a wide variety of proteins. It is essential for cell survival.
PRMT5 uses a cofactor, S-adenosyl-L-methionine (SAM), that is
converted to methylthioadenosine (MTA) during the reaction. In certain cancers
a gene called methylthioadenosine phosphorylase (MTAP) is deleted, causing an
accumulation of MTA and – through product inhibition – a decrease in PRMT5
activity. The idea is to develop a drug that binds to and further stabilizes the
(inactive) PRMT5•MTA complex, which is abundant in cancer cells, while not
interfering with the active form of the protein, which predominates in normal
cells. Told you it was complicated! [Note added: as befits the complicated biology I got a couple things wrong, corrected in the comment on 26 Jan.]
The researchers started with an
SPR screen of 1000 commercially available fragments, each at 100 µM. PRMT5 was
immobilized on the chip, with MTA added to the buffer to form the PRMT5•MTA
complex. This screen yielded 17 hits, and based on this encouraging result a
further set of nearly 1900 fragments was screened at 500 µM. The higher
concentration yielded significantly more hits, and when these were tested in
dose response experiments 100 were found with dissociation constants better
than 1 mM. The best 24 of these were then screened against PRMT5 loaded with
either MTA or the cofactor SAM. Compound F1 proved to be 5-fold selective for
the MTA-bound protein over the SAM-bound protein.
Crystallography revealed that
this molecule binds in the substrate-binding site in the vicinity of MTA and
suggested that it would clash with SAM binding, thus providing an explanation
for its selectivity. The crystal structure also revealed a nearby pocket that
could be targeted through fragment growing, and this was accomplished with
compound 2, which also showed activity in a biochemical assay. Further
structure-based design led eventually to compound 14, which was 26-fold
selective for the MTA-bound protein.
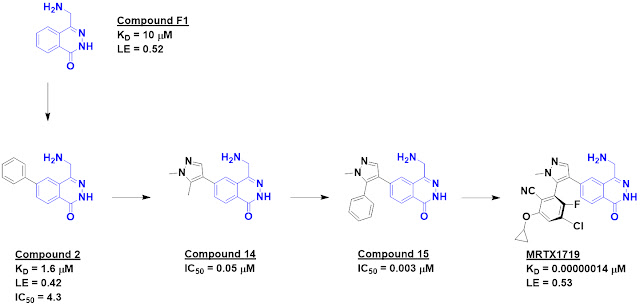
Crystallography revealed another
lipophilic pocket, and adding a phenyl group provided a nice increase in
potency in the form of compound 15. This molecule also showed low micromolar
cell activity. Further structure-based drug design ultimately led to MRTX1719;
the medicinal chemistry is elegant but beyond the scope of this post. Chemists
will recognize that the final molecule is an atropisomer. This type of
stereoisomer is uncommon in drugs in part because they can be difficult to
separate; the researchers note assessing 70 different conditions before
abandoning one series in favor of a more tractable one.
The dissociation constant of
MRTX1719 was measured by SPR as 0.14 pM and 9.4 pM for the PRMT5•MTA and
PRMT5•SAM complexes, respectively. We don’t encounter femtomolar binders very
often; the dissociation half-life for the MTA-bound protein is 14 days! The 67-fold
difference in binding was in good agreement with 70-80-fold differences in
cells without or with MTAP.
MRTX1719 was quite selective in a
panel of 42 methyltransferases. Pharmacokinetics and oral bioavailability were
good in mice, dogs, and cynomolgus monkeys. The molecule was well tolerated in
a mouse tumor model and caused tumor growth inhibition. Based on these results,
an IND for the molecule has been submitted to the FDA.
This is a lovely fragment-to-candidate
story, and Practical Fragments wishes everyone involved good fortune in
the clinic!


1 comment:
Correction brought to my attention by one of the researchers involved - thanks!
PRMT5 converts SAM to SAH, not MTA. MTA is a close analog of both and does mildly inhibit PRMT5. The synthetic lethality trick is that MTA is a substrate of MTAP which is often co-deleted with a cancer suppressor gene. Thus, MTA accumulates and we can leverage that difference in concentration to design selective inhibitors.
Post a Comment