Practical Fragments has
written previously about PhotoAffinity Bits, or PhABits, which are fragments
designed to reveal binding (as opposed to inhibition). These fragments contain
a photoreactive moiety such as a diazirine. When incubated with a protein
target and irradiated by ultraviolet light, the diazirine transforms into
reactive species that can react irreversibly with anything nearby. Intact
protein mass spectrometry can identify whether a reaction has occurred,
and further proteomics experiments can identify more precisely where the
PhABits reacted.
One challenge with this approach
is obtaining a library of PhABits; few are commercially available (though AstraZeneca
is sharing a set). In a recent open-access Chem. Sci. paper, Jacob Bush
and collaborators at GlaxoSmithKline and University of Strathclyde describe speeding
up the process.
The approach is called
direct-to-biology high-throughput chemistry (D2B-HTC). Recognizing that purification
is often the rate-limiting step in library synthesis, the researchers synthesized
PhABits in 384-well plates and used the crude reaction mixtures directly. In short,
a diazirine moiety linked to an activated ester was reacted for 24 hours with 1073
diverse alkylamines chosen from the GlaxoSmithKline internal collection. Interestingly,
54 of the amines themselves were not pure as judged by LC-MS – a useful
reminder of the importance of quality control. Ultimately, 853 of the reactions
were deemed successful, with >80% purity. Residual activated ester was quenched
with hydroxylamine, and the reactions were performed in biologically compatible
DMSO so that they could be used directly.
Next, each member of the library
was screened at 100 µM against human carbonic anhydrase I (CAI, at 1 µM), a
well-characterized model protein. After UV light illumination (302 nm for 10
minutes) the reactions were analyzed by mass spectrometry, resulting in seven
hits, defined as > 1.5% covalent adduct. Five of these contained a primary sulfonamide,
a privileged pharmacophore for carbonic anhydrases. Dose response experiments gave
similar results on both the crude mixtures as well as resynthesized, pure
compounds, with the best molecule showing high nanomolar activity. All seven PhABits
could be competed with the known ligand ethoxzolamide, suggesting that they
bind in the active site of the enzyme.
The seven hits gave even higher
levels of modification with carbonic anhydrase II (CAII), and four of the sulfonamide-containing
hits were further characterized by proteolyzing the modified enzyme and using
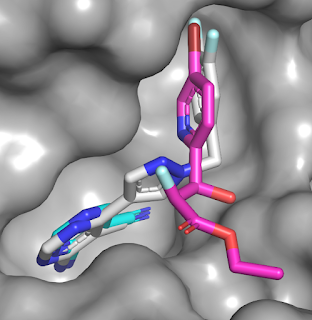
LC-MS/MS to determine the sites of modification. This revealed that the PhABits
were reacting with either a glutamic acid or histidine residue at the entrance
of the active site. As we discussed last year, the precise nature of the diazirine
probe can affect which amino acid residues are likely to react.
Based on the SAR from the primary
screen, a second 100-member library was constructed and screened without purification.
This provided a much higher hit rate, with all 52 hits containing a primary sulfonamide.
I do wish the researchers had
used an orthogonal method to assess the affinities of their molecules. One
drawback of the approach, which they note, is that “the absolute value of the
crosslinking yield is not indicative of binding affinity,” but it would be interesting
to know whether there is any correlation at all. It would also be nice to get a
sense of how often false negatives occur.
Still, D2B-HTC adds to the growing
list of methods that screen crude reaction mixtures, alongside related
approaches such as off-rate screening, Chemotype Evolution, and REFiLx.
The future may be a bit dirty, but perhaps we can get to our destination
faster.