If you’ve taken our poll on finding fragments you’ll have noticed ultrafiltration as one of the possibilities (and if you haven’t voted yet, please do so on the right-hand side of page). Ultrafiltration is grouped with affinity chromatography and capillary electrophoresis because all of these methods involve affinity-based separation of bound from unbound fragments. The technique is described in a recent paper in Anal. Bioanal. Chem.
The basic idea is simple: mix fragments with your protein of interest and then centrifuge through a membrane which retains large molecules such as proteins (and any bound fragments) but allows small molecules (unbound fragments) to pass through. If the composition of the filtrate differs from the composition of the initial mixture, one can assume that any depleted molecules are bound to the protein.
The researchers, all from the University of Washington, Seattle, have been using the technique on internal targets, and their recent paper gives a thorough account of how to do it and describes the results against two protein targets. In both cases, fragments were grouped into pools of 5 to 10 compounds, with each fragment at 0.098 mM and protein at 0.201 mM concentration; these conditions were chosen such that a 1 mM binder would give a 15% reduction in signal, which was roughly 3-fold over their standard deviation for control experiments. The ultrafiltration was done at 4 ˚C in 96-well plates, and the filtrates were analyzed by HPLC using a UV detector (all the fragments contained a chromophore, though the authors suggest that the experiment could also be done using mass spectrometry as a detector).
For the first protein, riboflavin kinase, the researchers found 4 hits out of 134 fragments tested, of which 3 confirmed as single compounds (ie, not in cocktails). Interestingly, these fragments were competed by the enzymatic product flavin mononucleotide but bound more tightly in the presence of the other product and cofactor, ADP and Mg(II). For the other protein, methionine aminopeptidase 1 from the parasite that causes malaria, 10 hits were found out of 243 fragments tested, of which 9 confirmed. The top 6 of these could be competed by methionine, the enzymatic product.
Overall this seems like an interesting method, though I do have one quibble: the authors do not report the activity of these fragments using other techniques. In theory it should be possible to extract dissociation constants from the % reduction in UV signal, and it would be very interesting to see how these values compare to dissociation constants measured using orthogonal methods. Has anyone out there done this?
15 September 2011
07 September 2011
Poll: fragment screening methods
Finding fragments is now routine, but how are people doing it? NMR of course has a venerable history, but X-ray crystallography provides higher resolution data, SPR is faster than either, and there are all sorts of other methods. To learn what’s state of the art (and to help me gather some data for an upcoming talk) please vote for what method(s) you’re using. Note that the vote is on the right side of the page, and you can vote for more than one method.
Affinity chromatography, capillary electrophoresis, or ultrafiltration
Computational screening
Functional screening (high concentration biochemical, FRET, etc.)
ITC (isothermal titration calorimetry)
MS (mass spectrometry)
NMR – ligand detected
NMR – protein detected
SPR (surface plasmon resonance)
Thermal shift assay
X-ray crystallography
Other – please specify in comments
Affinity chromatography, capillary electrophoresis, or ultrafiltration
Computational screening
Functional screening (high concentration biochemical, FRET, etc.)
ITC (isothermal titration calorimetry)
MS (mass spectrometry)
NMR – ligand detected
NMR – protein detected
SPR (surface plasmon resonance)
Thermal shift assay
X-ray crystallography
Other – please specify in comments
06 September 2011
Fragments vs BACE1: Amgen’s story
Some targets that have proven recalcitrant to standard screening approaches seem to be particularly amenable to fragment-based approaches. Beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) is one such example: Practical Fragments has previously discussed programs from both Evotec and Schering/Merck, the latter of which has resulted in more than one clinical candidate. In a recent issue of J. Med. Chem., researchers at Amgen describe their adventures with this Alzheimer’s disease target.
The researchers started by using SPR to screen a library of about 4000 fragments (which had MW < 300, polar surface area < 30 Å2, and ≤ 2 hydrogen bond donors). This led to 106 hits with 10 mM or better potency, of which 8 confirmed in an orthogonal assay with potency better than 1 mM. Among these was fragment 1, which was also discovered as a BACE1 binder by researchers at Astex using crystallographic screening.
 Astex’s crystallographic structure showed compound 1 packed pretty tightly into BACE1, but surprisingly, the Amgen team found that walking a bromine atom around the phenyl ring produced gains in potency at all four positions. In fact, adding an aromatic group off the 6-position, as in compound 34, led to a dramatic increase in potency, and crystallography revealed that the protein undergoes a conformational change to accommodate the extra bulk and form an edge-face interaction between a phenylalanine side chain and the added aromatic group.
Astex’s crystallographic structure showed compound 1 packed pretty tightly into BACE1, but surprisingly, the Amgen team found that walking a bromine atom around the phenyl ring produced gains in potency at all four positions. In fact, adding an aromatic group off the 6-position, as in compound 34, led to a dramatic increase in potency, and crystallography revealed that the protein undergoes a conformational change to accommodate the extra bulk and form an edge-face interaction between a phenylalanine side chain and the added aromatic group.
Putting substituents off the 3-position, as in compound 44, led to molecules that could access either the P1 pocket or the P2’ pocket of the enzyme, but adding the ortho-tolyl group from compound 34 to give compound 43 locked the binding mode down to the P2’ pocket and gave a satisfying boost in potency such that standard enzymatic assays could be used instead of SPR. Further medicinal chemistry led to picomolar binders such as compound 57 as well as compounds less active in the biochemical assay but with better permeability and lower efflux, such as compound 59. This compound also showed in vivo activity in a rat model, though it is rapidly metabolized.
Although crystallography was clearly enabling throughout the process, this paper is a warning not to be too slavish in adherence to structure, as the initial break (compound 1 to compound 34) would not have been predicted to be active based on the co-crystal structure of compound 1 with BACE1.
This is also another nice example of starting with a rather generic fragment (heck, one published by another group!) and advancing it to a potent, proprietary series.
The researchers started by using SPR to screen a library of about 4000 fragments (which had MW < 300, polar surface area < 30 Å2, and ≤ 2 hydrogen bond donors). This led to 106 hits with 10 mM or better potency, of which 8 confirmed in an orthogonal assay with potency better than 1 mM. Among these was fragment 1, which was also discovered as a BACE1 binder by researchers at Astex using crystallographic screening.

Putting substituents off the 3-position, as in compound 44, led to molecules that could access either the P1 pocket or the P2’ pocket of the enzyme, but adding the ortho-tolyl group from compound 34 to give compound 43 locked the binding mode down to the P2’ pocket and gave a satisfying boost in potency such that standard enzymatic assays could be used instead of SPR. Further medicinal chemistry led to picomolar binders such as compound 57 as well as compounds less active in the biochemical assay but with better permeability and lower efflux, such as compound 59. This compound also showed in vivo activity in a rat model, though it is rapidly metabolized.
Although crystallography was clearly enabling throughout the process, this paper is a warning not to be too slavish in adherence to structure, as the initial break (compound 1 to compound 34) would not have been predicted to be active based on the co-crystal structure of compound 1 with BACE1.
This is also another nice example of starting with a rather generic fragment (heck, one published by another group!) and advancing it to a potent, proprietary series.
29 August 2011
Fragment docking: it’s all about ligand efficiency
A widespread belief holds that it is more difficult to computationally dock fragment-sized molecules than lead-sized or drug-sized molecules. But is this really true? And if so, why? These questions are tackled by Marcel Verdonk and colleagues at Astex in a recent paper in J. Med. Chem.
The researchers examined 11 targets for which they had multiple crystal structures of each with bound fragments (which contained up to 15 non-hydrogen atoms) and larger molecules (which contained at least 20 non-hydrogen atoms); these crystal structures were the “correct” structures against which computational models could be judged. A total of 106 fragments and 100 larger molecules were then docked against their target proteins using a variety of different methods.
Surprisingly, the overall results were not overly impressive (<70% correct depending on methodology – often much less). But even more surprisingly, there was no difference between the success rates of the fragments and that of the larger molecules. However, the reasons for the poor performance were different. In the case of fragments, the problem was often that the scoring function didn’t recognize the correct solution; the energetics were just too subtle. In the case of the larger molecules, though, the problem was more often one of sampling: the docking program failed to produce the conformation of protein or ligand that corresponded to the correct solution, so it had no opportunity to score it. Potency made no difference: high-affinity compounds fared just as poorly as lower affinity compounds. What did make a difference, though, was ligand efficiency: compounds with high ligand-efficiency (> 0.4 kcal/mol/atom) were docked with considerably greater success than those with lower ligand efficiencies. As the authors point out, this makes sense intuitively:
The researchers examined 11 targets for which they had multiple crystal structures of each with bound fragments (which contained up to 15 non-hydrogen atoms) and larger molecules (which contained at least 20 non-hydrogen atoms); these crystal structures were the “correct” structures against which computational models could be judged. A total of 106 fragments and 100 larger molecules were then docked against their target proteins using a variety of different methods.
Surprisingly, the overall results were not overly impressive (<70% correct depending on methodology – often much less). But even more surprisingly, there was no difference between the success rates of the fragments and that of the larger molecules. However, the reasons for the poor performance were different. In the case of fragments, the problem was often that the scoring function didn’t recognize the correct solution; the energetics were just too subtle. In the case of the larger molecules, though, the problem was more often one of sampling: the docking program failed to produce the conformation of protein or ligand that corresponded to the correct solution, so it had no opportunity to score it. Potency made no difference: high-affinity compounds fared just as poorly as lower affinity compounds. What did make a difference, though, was ligand efficiency: compounds with high ligand-efficiency (> 0.4 kcal/mol/atom) were docked with considerably greater success than those with lower ligand efficiencies. As the authors point out, this makes sense intuitively:
High LE compounds form high-quality interactions with the target, which should make it easier for a docking program (both from a scoring and search perspective) to dock these compounds correctly.So the next time you see a computational model of a protein-ligand complex, you might want to take a closer look at ligand efficiency to get a sense of how trustworthy the structure might be.
25 August 2011
Journal of Computer-Aided Molecular Design 2011 Special FBDD Issue
The most recent issue of J. Comput. Aided Mol. Des. is entirely devoted to fragment-based drug discovery. This is the second special issue they’ve dedicated to this topic, the first one being in 2009.
Associate Editor Wendy Warr starts by interviewing Sandy Farmer of Boehringer Ingelheim. There are many insights and tips here, and I strongly recommend it for a view of how fragment-based approaches are practiced at one large company. A few quotes give a sense of the flavor.
On corporate environment:
Jean-Louis Reymond and colleagues have two articles for mining chemical structures, one analyzing their enumerated set of all compounds having up to 13 heavy atoms (GDB-13), the other focused on visualizing chemical space covered by molecules in PubChem. They have also put up a free web-based search tool (available here) for mining these databases.
Roland Bürli and colleagues at BioFocus describe their fragment library and its application to discover fragment hits against the kinase p38alpha. A range of techniques are used, with reasonably good correlation between them.
Finally, M. Catherine Johnson and colleagues present work they did at Pfizer on the anticancer target PDK1 (see here and here for other fragment-based approaches to this kinase). NMR screening provided a number of different fragment hits that were used to mine the corporate compound collection for more potent analogs, and crystallography-guided parallel chemistry ultimately led to low micromolar inhibitors.
Associate Editor Wendy Warr starts by interviewing Sandy Farmer of Boehringer Ingelheim. There are many insights and tips here, and I strongly recommend it for a view of how fragment-based approaches are practiced at one large company. A few quotes give a sense of the flavor.
On corporate environment:
In most cases, the difference between success and failure has little to do with the process and supporting technologies (they work!), but rather much more to do with the organizational structure to support FBDD and the organizational mindset to accept the different risk profile and resource model behind FBDD.On success rates:
We have found that FBDD has truly failed in only 2-3 targets out of over a dozen or so.On cost:
FBDD must be viewed as an investment opportunity, not a manufacturing process. And the business decisions surrounding FBDD should factor that in. FBDD is more about the opportunity cost (of not doing it) than the “run” cost (of doing it).On expertise:
Successful FBDD still requires a strong gut feeling.On small companies:
In the end, FBDD will always have a lower barrier to entry than HTS for a small company wanting to get into the drug-discovery space.There’s a lot of other really great content in the issue, much of which has been covered in previous posts on fragment library design, biolayer interferometry, LLEAT, and companies doing FBLD. The other articles are described briefly below.
The key to success for such companies is to identify or construct some technology platform.
Jean-Louis Reymond and colleagues have two articles for mining chemical structures, one analyzing their enumerated set of all compounds having up to 13 heavy atoms (GDB-13), the other focused on visualizing chemical space covered by molecules in PubChem. They have also put up a free web-based search tool (available here) for mining these databases.
Roland Bürli and colleagues at BioFocus describe their fragment library and its application to discover fragment hits against the kinase p38alpha. A range of techniques are used, with reasonably good correlation between them.
Finally, M. Catherine Johnson and colleagues present work they did at Pfizer on the anticancer target PDK1 (see here and here for other fragment-based approaches to this kinase). NMR screening provided a number of different fragment hits that were used to mine the corporate compound collection for more potent analogs, and crystallography-guided parallel chemistry ultimately led to low micromolar inhibitors.
21 August 2011
Designing fragment libraries
The topic of fragment library design is similar to foundation construction: most people don’t give it much thought, but any organization that doesn’t take it seriously could quickly find itself on shaky ground. Three recent papers cover different aspects of this topic.
The first paper, published in J. Comput. Aided Mol. Des. by researchers at Pfizer, describes the design and construction of their Global Fragment Initiative (GFI), a 2,885 fragment library meant to be broadly applicable to any target using any screening method (NMR, X-ray, SPR, MS, and biochemical assays). Most of these fragments came from commercial or in-house collections, but 293 were synthesized specifically for the library. All compounds were filtered to remove reactive or otherwise undesirable moieties. Interestingly, a large number of cationic and anionic molecules were included, based on the observation that many approved drugs are charged. Also, roughly a quarter of the compounds contained at least one chiral center.
Potential library members were put through a rather more rigorous selection than the standard Rule of 3 (for example, cLogP < 2.0). Molecular complexity was explicitly considered, and overly complex fragments were excluded. Fragments were also analyzed by 2D and 3D similarity and chosen to maximize diversity, though with the criterion that close analogs were available either in-house or commercially. Compounds were also chosen to allow rapid chemical elaboration. Finally, compounds were evaluated by NMR for purity and solubility at 1 mM in aqueous buffer and 50-100 mM in DMSO.
The bulk of the library (excluding custom-synthesized fragments) has been screened against at least 13 targets in 8 different protein families, mostly by NMR and biochemical assays, resulting in hit rates between 2.8 – 13%. Only one fragment hit all 13 targets, while 766 hit only one; in total 33% of the fragments hit one or more of the targets, a fraction eerily similar to that seen at Genentech and Vernalis. Overall this is a thorough, information-dense paper, and well worth reading if you are considering building or expanding a fragment library.
One of the most productive first steps you can take after identifying a fragment hit is to test close analogs or larger molecules that contain the fragment. Of course, it is easier to buy compounds than to make them, so a fragment library that effectively samples commercial compounds is likely to be useful. This “SAR by catalog” approach is the topic of the second paper, also in J. Comput. Aided Mol. Des., from Rod Hubbard and colleagues at Vernalis and the University of York.
The researchers analyzed catalogs of available compounds from each of three vendors (Asinex, Maybridge, and Specs). Filtering out undesirable functionalities and binning the molecules by size left 5600-7700 fragment-sized molecules and 28,600-252,000 larger molecules per vendor. Compound properties of the fragment sets (MW, polar surface area, number of hydrogen bond donors and acceptors, etc.) are summarized for each of the vendors, similarly to Chris Swain’s analysis. Six different algorithms were then tested to find sets of 200 fragments that would best represent the entire collection. In accordance with Murphy’s Law, the most complicated algorithm proved to be the most effective; it involves an iterative selection procedure with precisely defined similarity criteria. Still, this algorithm is not too difficult to implement, and it should prove a useful tool for selecting fragments from larger sets of commercial or in-house compounds.
Finally, a chapter by James Na and Qiyue Hu at Pfizer in a recent volume of Methods in Molecular Biology gives a broad overview of fragment library design. In addition to general considerations, the paper succinctly summarizes the design of the Pfizer Global Fragment Initiative as well as an earlier fragment library designed specifically for NMR screening. A more lengthy but instructive description of several Vernalis fragment libraries is also provided, as are some of the screening results. Finally, a nice table summarizes fragment libraries from more than a dozen companies.
The first paper, published in J. Comput. Aided Mol. Des. by researchers at Pfizer, describes the design and construction of their Global Fragment Initiative (GFI), a 2,885 fragment library meant to be broadly applicable to any target using any screening method (NMR, X-ray, SPR, MS, and biochemical assays). Most of these fragments came from commercial or in-house collections, but 293 were synthesized specifically for the library. All compounds were filtered to remove reactive or otherwise undesirable moieties. Interestingly, a large number of cationic and anionic molecules were included, based on the observation that many approved drugs are charged. Also, roughly a quarter of the compounds contained at least one chiral center.
Potential library members were put through a rather more rigorous selection than the standard Rule of 3 (for example, cLogP < 2.0). Molecular complexity was explicitly considered, and overly complex fragments were excluded. Fragments were also analyzed by 2D and 3D similarity and chosen to maximize diversity, though with the criterion that close analogs were available either in-house or commercially. Compounds were also chosen to allow rapid chemical elaboration. Finally, compounds were evaluated by NMR for purity and solubility at 1 mM in aqueous buffer and 50-100 mM in DMSO.
The bulk of the library (excluding custom-synthesized fragments) has been screened against at least 13 targets in 8 different protein families, mostly by NMR and biochemical assays, resulting in hit rates between 2.8 – 13%. Only one fragment hit all 13 targets, while 766 hit only one; in total 33% of the fragments hit one or more of the targets, a fraction eerily similar to that seen at Genentech and Vernalis. Overall this is a thorough, information-dense paper, and well worth reading if you are considering building or expanding a fragment library.
One of the most productive first steps you can take after identifying a fragment hit is to test close analogs or larger molecules that contain the fragment. Of course, it is easier to buy compounds than to make them, so a fragment library that effectively samples commercial compounds is likely to be useful. This “SAR by catalog” approach is the topic of the second paper, also in J. Comput. Aided Mol. Des., from Rod Hubbard and colleagues at Vernalis and the University of York.
The researchers analyzed catalogs of available compounds from each of three vendors (Asinex, Maybridge, and Specs). Filtering out undesirable functionalities and binning the molecules by size left 5600-7700 fragment-sized molecules and 28,600-252,000 larger molecules per vendor. Compound properties of the fragment sets (MW, polar surface area, number of hydrogen bond donors and acceptors, etc.) are summarized for each of the vendors, similarly to Chris Swain’s analysis. Six different algorithms were then tested to find sets of 200 fragments that would best represent the entire collection. In accordance with Murphy’s Law, the most complicated algorithm proved to be the most effective; it involves an iterative selection procedure with precisely defined similarity criteria. Still, this algorithm is not too difficult to implement, and it should prove a useful tool for selecting fragments from larger sets of commercial or in-house compounds.
Finally, a chapter by James Na and Qiyue Hu at Pfizer in a recent volume of Methods in Molecular Biology gives a broad overview of fragment library design. In addition to general considerations, the paper succinctly summarizes the design of the Pfizer Global Fragment Initiative as well as an earlier fragment library designed specifically for NMR screening. A more lengthy but instructive description of several Vernalis fragment libraries is also provided, as are some of the screening results. Finally, a nice table summarizes fragment libraries from more than a dozen companies.
17 August 2011
First fragment-based drug approved
Today marks history with the first FDA approval of a drug to come out of fragment-based screening. The drug is branded as Zelboraf (vemurafenib), but readers of this blog are probably more familiar with its previous name of PLX4032. Although widely expected to be approved, the FDA acted more than two months ahead of schedule. The drug targets a mutant form of BRAF and has received widespread media coverage because of dramatic clinical results showing that it extends life for patients with a particularly deadly form of skin cancer. FiercePharma has an article with links to several others.
The drug was discovered at Plexxikon and developed in partnership with Roche; Plexxikon was acquired earlier this year by Daiichi Sankyo. The PLX4032 story is a case study in how rapidly fragments can enable a program: initiated in Februrary 2005, it took just six years to reach approval. It’s also an example of starting with a profoundly unselective fragment and winding up with a very selective drug (see here for early discovery and here for characterization of PLX4032).

Although I claim no prescience, I did state back in 2008 that it would be nice if a fragment-based drug would be approved by 2011. But more importantly, it is worth pausing to remember that this is a victory not just for the field of fragment-based drug discovery, but for those patients afflicted with metastatic melanoma. In the end, that’s what this is all about.
The drug was discovered at Plexxikon and developed in partnership with Roche; Plexxikon was acquired earlier this year by Daiichi Sankyo. The PLX4032 story is a case study in how rapidly fragments can enable a program: initiated in Februrary 2005, it took just six years to reach approval. It’s also an example of starting with a profoundly unselective fragment and winding up with a very selective drug (see here for early discovery and here for characterization of PLX4032).

Although I claim no prescience, I did state back in 2008 that it would be nice if a fragment-based drug would be approved by 2011. But more importantly, it is worth pausing to remember that this is a victory not just for the field of fragment-based drug discovery, but for those patients afflicted with metastatic melanoma. In the end, that’s what this is all about.
12 August 2011
Fragment selectivity
A constant debate in fragment-based lead discovery is whether to focus on fragments that are selective for the target of interest. Because fragments have lower complexity than larger molecules they are likely to be less specific – that is, after all, one of the main arguments for why a small set of fragments can explore more chemical space than a much larger set of lead-like molecules. But does it make sense to prioritize those fragments that are more selective? In a recent issue of J. Med. Chem. Paul Bamborough and colleagues at GlaxoSmithKline address this question experimentally.
The broad family of kinases was chosen for the investigation. Protein kinases in particular have been a rich field for drug development, including fragment-based methods. The researchers assembled a library of 1065 commercially available fragments, most of which were designed to bind to the so-called “hinge” region of protein kinases where the substrate ATP binds. Of these fragments, 936 passed quality-control and maintained stability over the course of the year-plus study.
The researchers screened these fragments at 0.4 or 0.667 mM against a panel of 30 kinases using several different assay formats: FP (fluorescence polarization), IMAP (immobilization metal affinity phosphorylation), LEADseeker (a scintillation proximity assay), and TR-FRET (time-resolved fluorescence resonance energy transfer). Various experiments suggested that FP was most susceptible to assay artifacts, though the results were still usable.
17 of the fragments screened were chosen based on common fragment motifs in the literature. One example is adenine, a fragment of ATP, which of course is used by all kinases. Despite this universality, adenine actually showed surprising specificity, inhibiting some kinases strongly and not inhibiting others at all. The same goes for other hinge-binding fragments that we’ve seen before (such as indazole). On the other hand, biaryl urea fragments designed to bind to the less-conserved adaptive pocket of kinases were quite selective, hitting just 2 kinases strongly.
Of course, especially for kinases, the trick is not getting fragment hits but in figuring out which ones to pursue. Ligand efficiency is often used to prioritize fragments, but is this necessarily a good idea? The researchers compared published high-affinity inhibitors of several kinases with fragments contained within these inhibitors and found that the fragments often would not have stood out above the pack when compared solely on the basis of ligand efficiency. Even spookier, many of the most ligand-efficient fragments appear to be assay artifacts.
What about selectivity? Are non-selective fragments bound to become non-selective leads? The authors present one example of a rather non-selective fragment that could be optimized to a highly selective molecule; PLX4032, which started life as a promiscuous azaindole, is another example.
These are just anecdotes though, so to get a broader handle on this question the authors examined a set of 577 lead-like compounds that had been screened against 203 kinases. This led to a list of 592 matched pairs of lead-like compounds and fragment substructures (most of which are likely hinge-binders) which could be analyzed for selectivity. The results recapitulate a smaller, earlier study performed with a very different data set. As Bamborough et al. put it:
The broad family of kinases was chosen for the investigation. Protein kinases in particular have been a rich field for drug development, including fragment-based methods. The researchers assembled a library of 1065 commercially available fragments, most of which were designed to bind to the so-called “hinge” region of protein kinases where the substrate ATP binds. Of these fragments, 936 passed quality-control and maintained stability over the course of the year-plus study.
The researchers screened these fragments at 0.4 or 0.667 mM against a panel of 30 kinases using several different assay formats: FP (fluorescence polarization), IMAP (immobilization metal affinity phosphorylation), LEADseeker (a scintillation proximity assay), and TR-FRET (time-resolved fluorescence resonance energy transfer). Various experiments suggested that FP was most susceptible to assay artifacts, though the results were still usable.
17 of the fragments screened were chosen based on common fragment motifs in the literature. One example is adenine, a fragment of ATP, which of course is used by all kinases. Despite this universality, adenine actually showed surprising specificity, inhibiting some kinases strongly and not inhibiting others at all. The same goes for other hinge-binding fragments that we’ve seen before (such as indazole). On the other hand, biaryl urea fragments designed to bind to the less-conserved adaptive pocket of kinases were quite selective, hitting just 2 kinases strongly.
Of course, especially for kinases, the trick is not getting fragment hits but in figuring out which ones to pursue. Ligand efficiency is often used to prioritize fragments, but is this necessarily a good idea? The researchers compared published high-affinity inhibitors of several kinases with fragments contained within these inhibitors and found that the fragments often would not have stood out above the pack when compared solely on the basis of ligand efficiency. Even spookier, many of the most ligand-efficient fragments appear to be assay artifacts.
What about selectivity? Are non-selective fragments bound to become non-selective leads? The authors present one example of a rather non-selective fragment that could be optimized to a highly selective molecule; PLX4032, which started life as a promiscuous azaindole, is another example.
These are just anecdotes though, so to get a broader handle on this question the authors examined a set of 577 lead-like compounds that had been screened against 203 kinases. This led to a list of 592 matched pairs of lead-like compounds and fragment substructures (most of which are likely hinge-binders) which could be analyzed for selectivity. The results recapitulate a smaller, earlier study performed with a very different data set. As Bamborough et al. put it:
It is not uncommon to find selective lead-sized compounds based upon unselective fragments. Equally, unselective leadlike compounds are frequently based upon selective fragments. It seems that the property of selectivity need not be maintained between fragments and their related lead-sized molecules.On one level, Bamborough’s study is a bit discouraging: fragment selectivity should be used cautiously if at all in prioritizing fragments. Even ligand efficiency should not be gating; last year we discussed how a fragment with relatively modest ligand efficiency was transformed into the clinical-stage (and more ligand efficient) Hsp90 inhibitor AT13387. Other factors, such as structural novelty or how amenable a fragment will be to further elaboration, are just as if not more important for choosing fragments. All of which serves to reemphasize the fact that drug discovery is less a series of hard and fast rules than a loose system of guidelines and hunches. This lack of predictability is part of what makes the process so frustrating – and fun.
07 August 2011
Fragment-based events in 2011 and 2012
If you missed the fragment events earlier this year there is still one late addition to the calendar as well as some webinars. And it’s not too soon to be thinking about 2012!
August 16: Is your travel budget limited? Emerald Biosciences is putting together a series of free webinars related to FBLD on August 16, September 20, October 18, and November 15.
October 21: Zenobia Therapeutics is putting together a FBLD conference in San Diego. Although just one day, there is a nice lineup of speakers, so try to make it if you can.
2012
March 19-23: Keystone Symposium: Addressing the Challenges of Drug Discovery – Novel Targets, New Chemical Space and Emerging Approaches will be held in Tahoe City, CA. Although not exclusively devoted to fragments, there are many speakers I look forward to hearing.
April 17-19: Cambridge Healthtech Institute’s Seventh Annual Fragment-Based Drug Discovery will be held in San Diego. You can read impressions of this past year’s meeting here and 2010’s here.
September 23-26: FBLD 2012, the fourth in an illustrious series of conferences, will be held in my fair city of San Francisco. This should be a biggy – the first such event in the Bay Area (and the weather in September is usually decent too). You can read impressions of FBLD 2010 and FBLD 2009.
Know of anything else? Organizing a fragment event? Let us know and we’ll get the word out.
August 16: Is your travel budget limited? Emerald Biosciences is putting together a series of free webinars related to FBLD on August 16, September 20, October 18, and November 15.
October 21: Zenobia Therapeutics is putting together a FBLD conference in San Diego. Although just one day, there is a nice lineup of speakers, so try to make it if you can.
2012
March 19-23: Keystone Symposium: Addressing the Challenges of Drug Discovery – Novel Targets, New Chemical Space and Emerging Approaches will be held in Tahoe City, CA. Although not exclusively devoted to fragments, there are many speakers I look forward to hearing.
April 17-19: Cambridge Healthtech Institute’s Seventh Annual Fragment-Based Drug Discovery will be held in San Diego. You can read impressions of this past year’s meeting here and 2010’s here.
September 23-26: FBLD 2012, the fourth in an illustrious series of conferences, will be held in my fair city of San Francisco. This should be a biggy – the first such event in the Bay Area (and the weather in September is usually decent too). You can read impressions of FBLD 2010 and FBLD 2009.
Know of anything else? Organizing a fragment event? Let us know and we’ll get the word out.
03 August 2011
Ligand efficiency metrics poll results
Poll results are in, and not surprisingly, ligand efficiency (LE) comes out on top, with 86% of respondents using the metric. What was a surprise to me is how many folks use ligand lipophilic efficiency (LLE) (46%). Coming in a distant third at 15% is LLEAT, but given that this metric was just reported it has a pretty strong showing, and I wouldn't be surprised to see this increase. Binding efficiency index (BEI) comes in fourth with 12% of the vote, and Fsp3 is tied with "other" with 8% of the vote. The other metrics only received one or two votes each.
 Since people could vote on multiple metrics, there were more responses than respondents. Subtracting those who voted for "none" leaves 124 data points, suggesting that the average researcher is using 1.9 of these metrics (though unfortunately we don't have information on the median user).
Since people could vote on multiple metrics, there were more responses than respondents. Subtracting those who voted for "none" leaves 124 data points, suggesting that the average researcher is using 1.9 of these metrics (though unfortunately we don't have information on the median user).
Finally, for the 5 of you who selected "other", what else is out there that we've left out?
 Since people could vote on multiple metrics, there were more responses than respondents. Subtracting those who voted for "none" leaves 124 data points, suggesting that the average researcher is using 1.9 of these metrics (though unfortunately we don't have information on the median user).
Since people could vote on multiple metrics, there were more responses than respondents. Subtracting those who voted for "none" leaves 124 data points, suggesting that the average researcher is using 1.9 of these metrics (though unfortunately we don't have information on the median user).Finally, for the 5 of you who selected "other", what else is out there that we've left out?
30 July 2011
Fragments vs RNA revisited: the power of two
RNA can assume complex three-dimensional structures just like proteins, and given the many roles it plays it is perhaps surprising that there are so few drugs that target this class of biomolecules. One problem is that ribonucleic acids are less diverse than amino acids, so there is less scope for developing small molecules that bind to specific regions of RNA. Nonetheless, a few brave souls have tried, some using fragment-based approaches. The latest such effort appears in J. Mol. Biol.
The researchers, led by Gabriele Varani of the University of Washington, Seattle, took a two-step approach to find two fragments that could simultaneously bind to the TAR element of HIV-1, a short stem-loop element essential for viral replication. The protein that normally binds to TAR contains a critical arginine residue, so the researchers started by purchasing a set of 16 arginine mimetics and using NMR to determine if any of them bound to TAR RNA. Several did, and one guanidine-containing molecule (MV2003) gave a strong NMR signal and also contained a hydrophobic element. The researchers decided to use this to hunt for a second fragment.
To find the second binder, the team screened 250 generic (ie, not targeted to RNA) fragments from Maybridge in pools of 5-8 in the presence of the first fragment. Remarkably, saturation transfer difference (STD) experiments, which detect changes in ligand NMR signals upon binding to macromolecules, suggested that more than 100 of these generic fragments appeared to bind to TAR RNA. However, more careful study of 20 representative fragments from 13 different scaffolds rapidly winnowed the set: 5 didn’t repeat when tested outside the pool, 6 gave signals in the absence of RNA, and 3 were not dependent on the presence of MV2003, suggesting that they bind nonspecifically. However, the remaining 6 only produced signals in the presence of both TAR RNA and MV2003, indicating a specific ternary complex. Although two of these fragments contain a (positively charged) primary amine, the rest are likely either neutral or only partially protonated at physiological pH. Interestingly, one of these is closely related to an RNA-binding fragment identified in previous work by a different group.
Next, the researchers constructed a model of how MV2003 bound to RNA. They used NMR data (nuclear Overhauser effects, or NOEs) to determine which atoms of MV2003 were close to which atoms of TAR RNA. Unfortunately no intermolecular NOEs were observed between any of the six fragments and the RNA, but it was possible to observe interligand NOEs (ILOEs) between the fragments and MV2003, and this enabled additional modeling suggesting that the fragments bind in a small pocket that only forms when MV2003 binds to RNA.
The paper ends with a cliff-hanger:
The researchers, led by Gabriele Varani of the University of Washington, Seattle, took a two-step approach to find two fragments that could simultaneously bind to the TAR element of HIV-1, a short stem-loop element essential for viral replication. The protein that normally binds to TAR contains a critical arginine residue, so the researchers started by purchasing a set of 16 arginine mimetics and using NMR to determine if any of them bound to TAR RNA. Several did, and one guanidine-containing molecule (MV2003) gave a strong NMR signal and also contained a hydrophobic element. The researchers decided to use this to hunt for a second fragment.
To find the second binder, the team screened 250 generic (ie, not targeted to RNA) fragments from Maybridge in pools of 5-8 in the presence of the first fragment. Remarkably, saturation transfer difference (STD) experiments, which detect changes in ligand NMR signals upon binding to macromolecules, suggested that more than 100 of these generic fragments appeared to bind to TAR RNA. However, more careful study of 20 representative fragments from 13 different scaffolds rapidly winnowed the set: 5 didn’t repeat when tested outside the pool, 6 gave signals in the absence of RNA, and 3 were not dependent on the presence of MV2003, suggesting that they bind nonspecifically. However, the remaining 6 only produced signals in the presence of both TAR RNA and MV2003, indicating a specific ternary complex. Although two of these fragments contain a (positively charged) primary amine, the rest are likely either neutral or only partially protonated at physiological pH. Interestingly, one of these is closely related to an RNA-binding fragment identified in previous work by a different group.
Next, the researchers constructed a model of how MV2003 bound to RNA. They used NMR data (nuclear Overhauser effects, or NOEs) to determine which atoms of MV2003 were close to which atoms of TAR RNA. Unfortunately no intermolecular NOEs were observed between any of the six fragments and the RNA, but it was possible to observe interligand NOEs (ILOEs) between the fragments and MV2003, and this enabled additional modeling suggesting that the fragments bind in a small pocket that only forms when MV2003 binds to RNA.
The paper ends with a cliff-hanger:
The formation of a new binding pocket allows binding of other fragments and suggests that more powerful ligands can be generated by linking the fragments together.Although fragment linking is easier said than done, the hydrophobic moiety in MV2003 may improve the odds here, as described in the previous post. Practical Fragments hopes they will give it a shot!
25 July 2011
Fragment linking: oil and water do mix
Fragment linking is one of the most seductive forms of fragment-based lead discovery: take two low-affinity binders, link them together, and get a huge boost in potency. But what’s appealing in theory is difficult in practice: the linked molecule rarely binds more tightly than the product of the fragment affinities, and sometimes there is not even an improvement over the starting fragments. In a recent paper in Molecular Informatics, Mark Whittaker and colleagues at Evotec suggest a strategy to maximize the chance of success.
The researchers start by briefly reviewing nine published examples of fragment linking where affinities for both fragments as well the linked molecule are provided (some of these have been discussed previously here, here, and here). Of these, only three examples showed clear superadditivity (in which the linked molecule has a significantly higher affinity than the product of the affinities of the individual fragments), and two of these examples are rigged systems in which a molecule already known for its potency (such as biotin) is dissected into fragments. The challenges of linking are succinctly summarized:
All three of the examples that show superadditivity start with one fragment that is highly polar and makes hydrogen bonds or metal-mediated bonds with the protein. The researchers suggest that such fragments are likely to pay a heavy thermodynamic penalty when they are desolvated, and that this cost can be reduced by linking them to a hydrophobic fragment. Thus, to maximize your chances of successful linking, the authors suggest you should choose
This is an interesting proposal, though because there are so few examples it is hard to assess. Indeed, the only other case of clear superadditivity I found involves dimerizing a fragment that is reasonably hydrophobic (ClogP = 2.4), albeit negatively charged. Hopefully we’ll see more examples in the coming years, but in the meantime, linking a water-loving fragment to an oily one is worth a shot.
The researchers start by briefly reviewing nine published examples of fragment linking where affinities for both fragments as well the linked molecule are provided (some of these have been discussed previously here, here, and here). Of these, only three examples showed clear superadditivity (in which the linked molecule has a significantly higher affinity than the product of the affinities of the individual fragments), and two of these examples are rigged systems in which a molecule already known for its potency (such as biotin) is dissected into fragments. The challenges of linking are succinctly summarized:
The keys to achieving superadditivity upon linking are to maintain the binding modes of the parent fragments, not introduce both entropy and solvation penalties while designing the linker, and also make any interactions with the intervening protein surface that need to be made.Also, of course, the resulting molecule needs to be synthetically accessible. Having a certain amount of flexibility in the linker can be useful, as this will allow the fragments some room to shift around, but too much flexibility introduces an entropic cost that defeats the purpose of linking in the first place. Software tools such as those by BioSolveIT can help design the linker, but what if some fragments themselves are inherently better suited for linking?
All three of the examples that show superadditivity start with one fragment that is highly polar and makes hydrogen bonds or metal-mediated bonds with the protein. The researchers suggest that such fragments are likely to pay a heavy thermodynamic penalty when they are desolvated, and that this cost can be reduced by linking them to a hydrophobic fragment. Thus, to maximize your chances of successful linking, the authors suggest you should choose
a fragment pair that consists of one fragment that binds by strong H-bonds (or non-classical equivalents) and a second fragment that is more tolerant of changes in binding mode (hydrophobic or vdW binders).
This is an interesting proposal, though because there are so few examples it is hard to assess. Indeed, the only other case of clear superadditivity I found involves dimerizing a fragment that is reasonably hydrophobic (ClogP = 2.4), albeit negatively charged. Hopefully we’ll see more examples in the coming years, but in the meantime, linking a water-loving fragment to an oily one is worth a shot.
14 July 2011
Who's doing FBLD in 2011?
It’s been almost two years since our last attempt at cataloging companies doing fragment-based lead discovery. That list contained 19 entries, and 5 others were mentioned in the comments section. A paper just published online by Phil Hajduk and colleagues at Abbott in J. Comput. Aided Mol. Des. provides another list of 19 companies, which has inspired Practical Fragments to combine the two to provide what we hope is the most comprehensive list of companies working in FBLD. (The paper itself is worth reading too for insights into how fragment screening has evolved. For example, Abbott pioneered the use of NMR for finding and characterizing fragments, but now functional screening, modeling, and X-ray crystallography are dominant.)
Companies doing FBLD:
Abbott Laboratories
Ansaris (previously Locus)
AstraZeneca
Astex
Beactica
BioFocus (Galapagos)
BioLeap
Biosensor Tools
BioSolveIT
Boehringer Ingelheim
Carmot Therapeutics
Crelux
Crown Biosciences
Crystax Pharmaceuticals (web site seems down - are they still around?)
Eli Lilly
Emerald BioStructures (from deCODE)
Evotec
Genentech (Roche)
Genzyme (Sanfi-Aventis)
GlaxoSmithKline
Graffinity Pharmaceuticals (NovAliX)
iNovacia
IOTA Pharmaceuticals
Johnson & Johnson
Kinetic Discovery
MEDIT
Merck
Nerviano Medical Sciences
NovAliX
Novartis
Pfizer
Pharma Diagnostics
Plexxikon
Polyphor
Proteros
Pyxis Discovery
Roche
Schrodinger
Selcia
Sprint Bioscience
Structure Based Design
Vernalis
Zenobia Therapeutics
ZoBio
Unlike the previous list, this one also includes large pharmaceutical companies known to be active in FBLD. Companies that have been acquired or merged are listed separately if they maintain separate web sites. The list excludes companies solely focused on selling fragment libraries, as these are covered separately.
The current list includes some 44 companies, which illustrates how widespread FBLD has become. The continuity is also encouraging: despite the challenging economic environment of the last few years, aside from a few acquisitions, a name change, and a spin-off, all the companies from the 2009 list are still around (with the possible exception of Crystax).
I’m sure the list is still incomplete, so if you know of someone else please add them to the comment section.
Companies doing FBLD:
Abbott Laboratories
Ansaris (previously Locus)
AstraZeneca
Astex
Beactica
BioFocus (Galapagos)
BioLeap
Biosensor Tools
BioSolveIT
Boehringer Ingelheim
Carmot Therapeutics
Crelux
Crown Biosciences
Crystax Pharmaceuticals (web site seems down - are they still around?)
Eli Lilly
Emerald BioStructures (from deCODE)
Evotec
Genentech (Roche)
Genzyme (Sanfi-Aventis)
GlaxoSmithKline
Graffinity Pharmaceuticals (NovAliX)
iNovacia
IOTA Pharmaceuticals
Johnson & Johnson
Kinetic Discovery
MEDIT
Merck
Nerviano Medical Sciences
NovAliX
Novartis
Pfizer
Pharma Diagnostics
Plexxikon
Polyphor
Proteros
Pyxis Discovery
Roche
Schrodinger
Selcia
Sprint Bioscience
Structure Based Design
Vernalis
Zenobia Therapeutics
ZoBio
Unlike the previous list, this one also includes large pharmaceutical companies known to be active in FBLD. Companies that have been acquired or merged are listed separately if they maintain separate web sites. The list excludes companies solely focused on selling fragment libraries, as these are covered separately.
The current list includes some 44 companies, which illustrates how widespread FBLD has become. The continuity is also encouraging: despite the challenging economic environment of the last few years, aside from a few acquisitions, a name change, and a spin-off, all the companies from the 2009 list are still around (with the possible exception of Crystax).
I’m sure the list is still incomplete, so if you know of someone else please add them to the comment section.
07 July 2011
Biolayer interferometry (BLI)
Surface plasmon resonance (SPR) has become a primary tool for finding fragments. One of its attractions is that, in addition to requiring only small amounts of protein, it can provide dissociation constants (Kd values) and, for tighter binders, on-rates and off-rates. However, SPR is not the only biosensor-based technology out there. Biolayer interferometry is a related technique, and, as judged by the discussion following the FBLD 2010 meeting, is clearly of interest to many people. A paper published online by Charles Wartchow and colleagues in J. Comput. Aided Mol. Des. provides a description of the technology and comparison with other methods.
Like SPR, BLI requires immobilization of the protein target to a surface; the current paper uses biotin-labeled proteins and streptavidin coated biosensors from ForteBio. Unlike SPR, the technology does not rely on samples flowing through tiny capillaries, and up to 16 protein-labeled sensors can be simultaneously dipped directly into different solutions of small molecules arrayed in a 384-well plate. BLI relies on changes in the interference pattern of light between the sensor and the solution caused when a small molecule binds to a protein on the surface of the sensor.
In the current study, the authors studied three proteins: Bcl-2, JNK1, and eIF4E. Initially a library of 140 fragments was screened in triplicate at 200 micromolar concentration against each of the three targets. Both JNK1 and Bcl-2 gave very high hit rates (24 and 21%, respectively), but eIF4E gave a much more “fragment typical” hit rate of 3.5%. This protein was subsequently screened against 6500 compounds, a task which required 1 mg of protein, 10 days, and 700 sensors (which needed to be periodically replaced throughout the campaign).
After curating the eIF4E hits to remove compounds that gave anomalously high signals or slow off-rates, the remaining molecules were then retested in a second screen, which confirmed 50% of the remaining hits, for an overall hit rate of 1.3%. However, many of these still looked suspicious when they were tested in 8-point titration curves; it seems that, like SPR, BLI is also prone to false-positive problems.
The researchers also ran biochemical and SPR screens on some of the targets. For eIF4E, the overlap between hits coming from BLI and those from biochemical screens was 52%, though many of these are derivatives of a single scaffold. Another subset of the common hits gave non-ideal behavior, calling into question their mechanism of action. It remains unclear whether the BLI hits that were not active in biochemical assays are real, and if so, relevant.
In the end, the authors conclude that:
These fragment screening studies demonstrate that BLI is suitable for small molecule characterization and fragment screening.
But they continue:
Hit assessment… with BLI and SPR is non-trivial, however, and although numerous hits from the BLI, SPR, and biochemical assays were characterized, most of the BLI and SPR data obtained from the examination of a concentration series in the micromolar range showed linear relationships with respect to concentration, unreasonably high signals, or slow off-rates.
Clearly, like all techniques, one should not rely on BLI alone. What remains to be seen is whether BLI has advantages over related techniques such as SPR, whether in terms of speed, sensitivity, resistance to artifacts, or cost. Several of the authors of the paper are from Roche, but the paper does not make clear whether BLI is becoming integrated into the workflow there. Is anyone else out there using BLI? If so, what has been your experience?
Like SPR, BLI requires immobilization of the protein target to a surface; the current paper uses biotin-labeled proteins and streptavidin coated biosensors from ForteBio. Unlike SPR, the technology does not rely on samples flowing through tiny capillaries, and up to 16 protein-labeled sensors can be simultaneously dipped directly into different solutions of small molecules arrayed in a 384-well plate. BLI relies on changes in the interference pattern of light between the sensor and the solution caused when a small molecule binds to a protein on the surface of the sensor.
In the current study, the authors studied three proteins: Bcl-2, JNK1, and eIF4E. Initially a library of 140 fragments was screened in triplicate at 200 micromolar concentration against each of the three targets. Both JNK1 and Bcl-2 gave very high hit rates (24 and 21%, respectively), but eIF4E gave a much more “fragment typical” hit rate of 3.5%. This protein was subsequently screened against 6500 compounds, a task which required 1 mg of protein, 10 days, and 700 sensors (which needed to be periodically replaced throughout the campaign).
After curating the eIF4E hits to remove compounds that gave anomalously high signals or slow off-rates, the remaining molecules were then retested in a second screen, which confirmed 50% of the remaining hits, for an overall hit rate of 1.3%. However, many of these still looked suspicious when they were tested in 8-point titration curves; it seems that, like SPR, BLI is also prone to false-positive problems.
The researchers also ran biochemical and SPR screens on some of the targets. For eIF4E, the overlap between hits coming from BLI and those from biochemical screens was 52%, though many of these are derivatives of a single scaffold. Another subset of the common hits gave non-ideal behavior, calling into question their mechanism of action. It remains unclear whether the BLI hits that were not active in biochemical assays are real, and if so, relevant.
In the end, the authors conclude that:
These fragment screening studies demonstrate that BLI is suitable for small molecule characterization and fragment screening.
But they continue:
Hit assessment… with BLI and SPR is non-trivial, however, and although numerous hits from the BLI, SPR, and biochemical assays were characterized, most of the BLI and SPR data obtained from the examination of a concentration series in the micromolar range showed linear relationships with respect to concentration, unreasonably high signals, or slow off-rates.
Clearly, like all techniques, one should not rely on BLI alone. What remains to be seen is whether BLI has advantages over related techniques such as SPR, whether in terms of speed, sensitivity, resistance to artifacts, or cost. Several of the authors of the paper are from Roche, but the paper does not make clear whether BLI is becoming integrated into the workflow there. Is anyone else out there using BLI? If so, what has been your experience?
30 June 2011
How effectively can fragments sample chemical space?
One of the key advantages of fragment-based drug discovery is that, since there are fewer fragments than lead-sized or drug-sized molecules, it is possible to sample chemical space far more efficiently with fragments than with larger molecules. At least, that’s the theory, but does is it hold true in the real world?
To put it another way, do fragments sample all of the space in which drugs are found? And what kinds of fragments are best for this sampling? In the most recent issue of J. Med. Chem., Stephen Roughley and Rod Hubbard of Vernalis address such questions.
The system they investigate, heat shock protein 90 (Hsp90), is an ideal model system: it is both a popular anti-cancer target as well as structurally tractable, and is thus arguably the most heavily explored single target in terms of fragment-based lead discovery. At least 8 antagonists have entered the clinic, of which at least 2 have come from fragments (see the posts on AT13387, NVP-BEP800/VER-82576, and posts on Evotec compounds discovered by fragment growing or linking.)
Vernalis has had a long-running fragment-based program targeting Hsp90, which has resulted in numerous fragments whose binding modes have been determined by X-ray crystallography. Roughley and Hubbard analyzed these fragments and compared them to published inhibitors. Just 5 distinct fragments can be mapped onto all of the clinical compounds: a handful of fragments effectively samples relevant chemical space. As the authors put it:
For Hsp90 at least, the fragments do cover an appropriate chemical space; what is then important is the imagination of the chemist in evolving the fragments into potent inhibitors.
The second point – about the imagination of the chemist – is critical. Mapping fragments onto elaborated molecules is easier to do retrospectively than prospectively; a cynic could argue that methane is a fragment of just about any drug out there. However, Roughly and Hubbard also point out that, particularly in cases such as this where there are many co-crystal structures, fragments can help identify bioisosteres, including cryptic ones that would not be obvious purely from studying functional SAR.
The paper also addresses the issue of optimal library design, in particular the dilemma of size. Although all five representative fragments were found in an initial set of just 719 fragments, subtle changes can dramatically change the binding mode, an issue we’ve touched on previously. It may not be practical to have multiple similar fragments present in a primary screening library, but testing close analogs after identifying initial fragment hits is likely to be worthwhile.
Finally, one of the concerns about fragment-based approaches is that, if everyone is buying the same set of fragments from the same suppliers and screening them against the same targets, they will end up in the same place – and stumbling over each others’ intellectual property. Reassuringly, this turns out not to be the case:
Even though the various companies discovered rather similar compounds from a fragment screen, exploiting similar binding motifs, there were no exact matches. [Also], the subsequent evolution of the fragments sometimes took very different paths and produced mostly very different chemical leads and candidates.
If this holds true for such heavily mined targets as Hsp90 (and kinases, as discussed previously) it should be even more true for newer classes of targets.
There is a wealth of information in this paper, and it is worth perusing, especially if you find yourself longing for some science over the long holiday weekend.
To put it another way, do fragments sample all of the space in which drugs are found? And what kinds of fragments are best for this sampling? In the most recent issue of J. Med. Chem., Stephen Roughley and Rod Hubbard of Vernalis address such questions.
The system they investigate, heat shock protein 90 (Hsp90), is an ideal model system: it is both a popular anti-cancer target as well as structurally tractable, and is thus arguably the most heavily explored single target in terms of fragment-based lead discovery. At least 8 antagonists have entered the clinic, of which at least 2 have come from fragments (see the posts on AT13387, NVP-BEP800/VER-82576, and posts on Evotec compounds discovered by fragment growing or linking.)
Vernalis has had a long-running fragment-based program targeting Hsp90, which has resulted in numerous fragments whose binding modes have been determined by X-ray crystallography. Roughley and Hubbard analyzed these fragments and compared them to published inhibitors. Just 5 distinct fragments can be mapped onto all of the clinical compounds: a handful of fragments effectively samples relevant chemical space. As the authors put it:
For Hsp90 at least, the fragments do cover an appropriate chemical space; what is then important is the imagination of the chemist in evolving the fragments into potent inhibitors.
The second point – about the imagination of the chemist – is critical. Mapping fragments onto elaborated molecules is easier to do retrospectively than prospectively; a cynic could argue that methane is a fragment of just about any drug out there. However, Roughly and Hubbard also point out that, particularly in cases such as this where there are many co-crystal structures, fragments can help identify bioisosteres, including cryptic ones that would not be obvious purely from studying functional SAR.
The paper also addresses the issue of optimal library design, in particular the dilemma of size. Although all five representative fragments were found in an initial set of just 719 fragments, subtle changes can dramatically change the binding mode, an issue we’ve touched on previously. It may not be practical to have multiple similar fragments present in a primary screening library, but testing close analogs after identifying initial fragment hits is likely to be worthwhile.
Finally, one of the concerns about fragment-based approaches is that, if everyone is buying the same set of fragments from the same suppliers and screening them against the same targets, they will end up in the same place – and stumbling over each others’ intellectual property. Reassuringly, this turns out not to be the case:
Even though the various companies discovered rather similar compounds from a fragment screen, exploiting similar binding motifs, there were no exact matches. [Also], the subsequent evolution of the fragments sometimes took very different paths and produced mostly very different chemical leads and candidates.
If this holds true for such heavily mined targets as Hsp90 (and kinases, as discussed previously) it should be even more true for newer classes of targets.
There is a wealth of information in this paper, and it is worth perusing, especially if you find yourself longing for some science over the long holiday weekend.
26 June 2011
Ligand efficiency and related metrics (Poll)
The discussion following the recent post on LLEAT got me thinking that metrics could be a good topic for a poll (see right side of page).
Click below to see definitions or references, and vote on the right side of the page. Note that you can choose multiple answers.
Antibacterial efficiency
Binding efficiency index (BEI)
Fit quality (FQ)
Fsp3
Ligand efficiency (LE)
Ligand-efficiency-dependent lipophilicity (LELP)
Ligand lipophilic efficiency (LLE)
LLEAT
%LE
Percentage efficiency index (PEI)
Surface-binding efficiency index (SEI)
Other
None
Also, if you use any additional metrics or want to be more specific about how or why you use (or don’t use) the above metrics, please comment.
Click below to see definitions or references, and vote on the right side of the page. Note that you can choose multiple answers.
Antibacterial efficiency
Binding efficiency index (BEI)
Fit quality (FQ)
Fsp3
Ligand efficiency (LE)
Ligand-efficiency-dependent lipophilicity (LELP)
Ligand lipophilic efficiency (LLE)
LLEAT
%LE
Percentage efficiency index (PEI)
Surface-binding efficiency index (SEI)
Other
None
Also, if you use any additional metrics or want to be more specific about how or why you use (or don’t use) the above metrics, please comment.
20 June 2011
Ligand Lipophilicity Efficiency AT Astex Therapeutics
Our last post discussed the growing plague of molecular obesity, and how numerous metrics have been designed to control it. In a paper published online in J. Comput. Aided Mol. Des. Paul Mortenson and Chris Murray of Astex describe a new one: LLEAT.
Although ligand efficiency (LE) is probably the most widely used and intuitive metric, it does not take into account lipophilicity. Other indices do, notably ligand lipophilicity efficiency (LLE) and ligand-efficiency-dependent lipophilicity (LELP), but these both have drawbacks for evaluating fragments. LLE (defined as pIC50 – log P) is not normalized for size; for a fragment to have an (attractive) LLE ≥ 5 it would need an exceptionally low log P or an exceptionally high affinity. LELP, defined as log P / ligand efficiency, is also potentially misleading since a compound could have an acceptable LELP value even with a low ligand efficiency if the log P is also very low.
To address these problems, Mortenson and Murray have tried to strip out the non-specific binding a lipophilic molecule experiences when going from water to a binding site in a protein. They define this modified free energy of binding as:
ΔG* = ΔG - ΔGlipo
≈ RT ln (IC50) + RT ln (P)
≈ ln (10) * RT (log P - pIC50)
In order to put values coming out of this metric on the same scale as those from ligand efficiency, they add a constant, such that:
LLEAT = 0.11 – ΔG* / (number of heavy atoms)
Thus, just as in ligand efficiency, the goal is for molecules to have LLEAT ≥ 0.3 kcal/mol per heavy atom.
The index has some interesting implications. For example, the two fragments below have the same number of heavy atoms, and thus if they had the same activity they would have the same ligand efficiency; on this measure alone, neither would be preferred as a starting point for further work. However, because of their very different lipophilicities, fragment 2 would need to be 45 times more potent than fragment 1 in order to have the same LLEAT of at least 0.3.
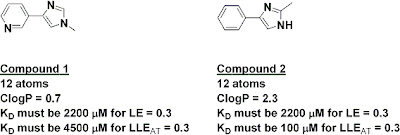 A similar analysis can be done during optimization. For example, adding either a phenyl or a piperazinyl substituent should produce a 20-fold boost in potency in order to maintain ligand efficiency at 0.3, since both have 6 atoms. However, in order to maintain LLEAT at 0.3, the phenyl would need to produce a 460-fold boost in potency while the piperazinyl would need to improve potency only 3-fold. This is consistent with what other folks have reported qualitatively, but it’s nice to have a simple quantitative measure.
A similar analysis can be done during optimization. For example, adding either a phenyl or a piperazinyl substituent should produce a 20-fold boost in potency in order to maintain ligand efficiency at 0.3, since both have 6 atoms. However, in order to maintain LLEAT at 0.3, the phenyl would need to produce a 460-fold boost in potency while the piperazinyl would need to improve potency only 3-fold. This is consistent with what other folks have reported qualitatively, but it’s nice to have a simple quantitative measure.
Although some people may groan at yet another index, and no metric is perfect, I like the fact that this one is intuitive and has the same range of “acceptable” values as ligand efficiency. What do you think – is it useful?
Although ligand efficiency (LE) is probably the most widely used and intuitive metric, it does not take into account lipophilicity. Other indices do, notably ligand lipophilicity efficiency (LLE) and ligand-efficiency-dependent lipophilicity (LELP), but these both have drawbacks for evaluating fragments. LLE (defined as pIC50 – log P) is not normalized for size; for a fragment to have an (attractive) LLE ≥ 5 it would need an exceptionally low log P or an exceptionally high affinity. LELP, defined as log P / ligand efficiency, is also potentially misleading since a compound could have an acceptable LELP value even with a low ligand efficiency if the log P is also very low.
To address these problems, Mortenson and Murray have tried to strip out the non-specific binding a lipophilic molecule experiences when going from water to a binding site in a protein. They define this modified free energy of binding as:
ΔG* = ΔG - ΔGlipo
≈ RT ln (IC50) + RT ln (P)
≈ ln (10) * RT (log P - pIC50)
In order to put values coming out of this metric on the same scale as those from ligand efficiency, they add a constant, such that:
LLEAT = 0.11 – ΔG* / (number of heavy atoms)
Thus, just as in ligand efficiency, the goal is for molecules to have LLEAT ≥ 0.3 kcal/mol per heavy atom.
The index has some interesting implications. For example, the two fragments below have the same number of heavy atoms, and thus if they had the same activity they would have the same ligand efficiency; on this measure alone, neither would be preferred as a starting point for further work. However, because of their very different lipophilicities, fragment 2 would need to be 45 times more potent than fragment 1 in order to have the same LLEAT of at least 0.3.
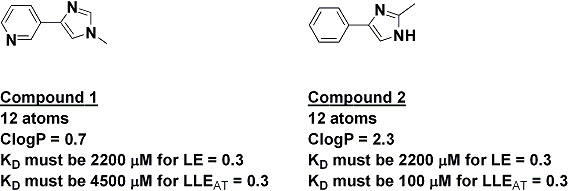
Although some people may groan at yet another index, and no metric is perfect, I like the fact that this one is intuitive and has the same range of “acceptable” values as ligand efficiency. What do you think – is it useful?
12 June 2011
Beware molecular obesity
Obesity in humans is a growing problem, and not just aesthetically: the condition may be responsible for millions of premature deaths. In a recent article in Med. Chem. Commun., Mike Hann of GlaxoSmithKline notes that “molecular obesity” is also leading to the untimely demise of far too many drug development programs.
Hann, who is especially known for his work on molecular complexity, defines molecular obesity as the “tendency to build potency into molecules by the inappropriate use of lipohilicity.” This is the result of an unhealthy “addiction” to potency. Hann suggests that since potency is easy to measure it is pursued preferentially to other factors, particularly early in a program. This is all too often achieved by adding mass, much of it lipophilic. The problem is that all this grease decreases solubility and increases the risks of off-target binding and toxic side effects.
Tools such as the Rule of 5 have been developed in part to avoid this problem, and a number of other indices have been introduced more recently. For example, lipophilic ligand efficiency (LLE) is defined as pIC50 – LogP; molecules with an LLE > 5 are likely to be more developable. Other guidelines that Hann mentions and that have been covered here include LELP, number of aromatic rings, and fraction of sp3 hybridized carbon atoms. But this is not to say that metrics will save the day:
Hann also argues that potency itself is over-rated: many teams seek single digit nanomolar binders even though approved drugs have average potencies of 20 nM to 200 nM.
The paper is a fun read (and is free after registration too). Also, Hann one-ups Donald Rumsfield’s (in)famous “known knowns, known unknowns, and unknown unknowns” by pointing out that much of this information falls into the category of “unknown knowns”:
Hann, who is especially known for his work on molecular complexity, defines molecular obesity as the “tendency to build potency into molecules by the inappropriate use of lipohilicity.” This is the result of an unhealthy “addiction” to potency. Hann suggests that since potency is easy to measure it is pursued preferentially to other factors, particularly early in a program. This is all too often achieved by adding mass, much of it lipophilic. The problem is that all this grease decreases solubility and increases the risks of off-target binding and toxic side effects.
Tools such as the Rule of 5 have been developed in part to avoid this problem, and a number of other indices have been introduced more recently. For example, lipophilic ligand efficiency (LLE) is defined as pIC50 – LogP; molecules with an LLE > 5 are likely to be more developable. Other guidelines that Hann mentions and that have been covered here include LELP, number of aromatic rings, and fraction of sp3 hybridized carbon atoms. But this is not to say that metrics will save the day:
The problem with the proliferation of so many “rules” is the trend to slavishly apply them without really understanding their required context for use and subsequent limitations.Starting a program with the smallest possible lead should in theory lead to smaller drugs, and this is one of the key justifications for fragment-based approaches, though even here it is important that the molecules do not become obese during optimization. One of the themes at the 6th annual FBDD conference was to do a bit of optimization around the fragment itself before growing or linking. On a related note, Rod Hubbard warned in his opening presentation at the conference to “beware the super-sized fragment.” Not only are larger fragments likely to be less complementary to the target, the number of possibilities increases (and thus the coverage of chemical space drops) by roughly ten-fold with each atom added to a fragment.
Hann also argues that potency itself is over-rated: many teams seek single digit nanomolar binders even though approved drugs have average potencies of 20 nM to 200 nM.
The paper is a fun read (and is free after registration too). Also, Hann one-ups Donald Rumsfield’s (in)famous “known knowns, known unknowns, and unknown unknowns” by pointing out that much of this information falls into the category of “unknown knowns”:
Those things that are known but have become unknown, either because we have never learnt them, or forgotten about them, or more dangerously chosen to ignoreThis review is an excellent corrective to the first two problems, and a clear warning about the third.
30 May 2011
Perverse trade-offs: the maximal enthalpy of ligands
Most readers of this blog are familiar with the concept of ligand efficiency:
LE = (free energy of ligand binding) / (number of heavy atoms)
The metric is easy to calculate, intuitive, and useful for evaluating fragments. Weirdly, as ligands get larger, the ligand efficiencies tend to decrease. This is a consequence of the fact that the maximal affinities of ligands also start to plateau as their sizes increase, a fact noted more than a decade ago by Kuntz and colleagues. The reason for this has never been satisfactorily explained, but the effect is so pronounced that some researchers have proposed alternatives to LE that take it into account, for example %LE and fit quality. In a paper published online in ACS Med. Chem. Lett., Charles Reynolds and M. Katherine Holloway have delved into the origins of declining LE with increasing size in more detail.
The researchers analyzed 102 ligand-protein complexes representing 14 target classes for which thermodynamic data had been collected using isothermal titration calorimetry (ITC). They calculated ligand efficiency as well as “enthalpy efficiency” and “entropy efficiency”, where enthalpy or entropy takes the place of free energy in the numerator. In the case of enthalpy efficiency, the trend was the same as for ligand efficiency: as molecules got larger, the enthalpy efficiencies tended to decrease. For entropy efficiencies, however, there was essentially no trend. In other words, the leveling out of binding energy is due to enthalpic effects more than to entropic effects. The authors suggest that:
The size effect on enthalpy is a result of increasing ligand complexity and the need to satisfy multiple geometric constraints simultaneously.
The findings thus provide further support for trying to identify fragments whose binding is largely enthalpically driven, but, as usual in the real world, things are not quite so easy. One of the frustrating things about medicinal chemistry is the phenomenon of enthalpy-entropy compensation: if you try to improve the enthalpy of binding, say by adding a hydrogen-bond acceptor, you may end up paying an entropic penalty such that overall binding increases only modestly, if at all. Indeed, the data in the paper show a very strong correlation between enthalpy and entropy: enthalpic binders tend to have negative (unfavorable) entropy, and the most entropic binders actually have positive (highly unfavorable) enthalpies of binding.
Worse, despite the striking correlation between enthalpy and entropy, there is almost no correlation between free energy of binding and enthalpy or entropy. The researchers suggest that these perverse trends (or lack thereof) explain why computational approaches to drug discovery are not more successful: most modeling relies on one or a few low-energy conformations, in effect ignoring entropy. However, there is a correlation between overall binding energy and enthalpy for certain targets, such as HIV protease and aldose reductase; perhaps such targets are more amenable to modeling.
In summary, the message seems to be that you should try to find enthalpic binders if you can, though the binding energetics may change unpredictably during optimization. For modelers, the research suggests that ignoring entropy is unwise. For medicinal chemists, I’m not sure how much of this is actionable, but sometimes it’s nice to know the origins behind the perversity of the universe.
LE = (free energy of ligand binding) / (number of heavy atoms)
The metric is easy to calculate, intuitive, and useful for evaluating fragments. Weirdly, as ligands get larger, the ligand efficiencies tend to decrease. This is a consequence of the fact that the maximal affinities of ligands also start to plateau as their sizes increase, a fact noted more than a decade ago by Kuntz and colleagues. The reason for this has never been satisfactorily explained, but the effect is so pronounced that some researchers have proposed alternatives to LE that take it into account, for example %LE and fit quality. In a paper published online in ACS Med. Chem. Lett., Charles Reynolds and M. Katherine Holloway have delved into the origins of declining LE with increasing size in more detail.
The researchers analyzed 102 ligand-protein complexes representing 14 target classes for which thermodynamic data had been collected using isothermal titration calorimetry (ITC). They calculated ligand efficiency as well as “enthalpy efficiency” and “entropy efficiency”, where enthalpy or entropy takes the place of free energy in the numerator. In the case of enthalpy efficiency, the trend was the same as for ligand efficiency: as molecules got larger, the enthalpy efficiencies tended to decrease. For entropy efficiencies, however, there was essentially no trend. In other words, the leveling out of binding energy is due to enthalpic effects more than to entropic effects. The authors suggest that:
The size effect on enthalpy is a result of increasing ligand complexity and the need to satisfy multiple geometric constraints simultaneously.
The findings thus provide further support for trying to identify fragments whose binding is largely enthalpically driven, but, as usual in the real world, things are not quite so easy. One of the frustrating things about medicinal chemistry is the phenomenon of enthalpy-entropy compensation: if you try to improve the enthalpy of binding, say by adding a hydrogen-bond acceptor, you may end up paying an entropic penalty such that overall binding increases only modestly, if at all. Indeed, the data in the paper show a very strong correlation between enthalpy and entropy: enthalpic binders tend to have negative (unfavorable) entropy, and the most entropic binders actually have positive (highly unfavorable) enthalpies of binding.
Worse, despite the striking correlation between enthalpy and entropy, there is almost no correlation between free energy of binding and enthalpy or entropy. The researchers suggest that these perverse trends (or lack thereof) explain why computational approaches to drug discovery are not more successful: most modeling relies on one or a few low-energy conformations, in effect ignoring entropy. However, there is a correlation between overall binding energy and enthalpy for certain targets, such as HIV protease and aldose reductase; perhaps such targets are more amenable to modeling.
In summary, the message seems to be that you should try to find enthalpic binders if you can, though the binding energetics may change unpredictably during optimization. For modelers, the research suggests that ignoring entropy is unwise. For medicinal chemists, I’m not sure how much of this is actionable, but sometimes it’s nice to know the origins behind the perversity of the universe.
19 May 2011
Fragments vs Pim-1
The three members of the Pim family of serine/threonine kinases help cancer cells proliferate and survive, and are thus interesting as potential anti-cancer targets. Structurally the kinases are also intriguing because they have a proline residue in the “hinge” region of the purine binding site, differentiating them from the other 500+ kinases in the human genome. Two recent papers describe different fragment-screening approaches against Pim-1: one case rediscovers fragments of a previously reported compound and the other identifies a new series of potent inhibitors unlike other reported kinase inhibitors.
In the first paper, published in Acta Cryst. Section D, researchers from Bayer Schering Pharma AG performed a crystallographic screen of just 36 fragments against Pim-1. Despite the small library size, a dozen of the fragments produced interpretable electron density, although only 4 of these showed activity better than 10 mM. Fragment 3, with an IC50 of 130 micromolar in an enzymatic assay, was the most potent. Interestingly, a close analog of this cinnamic acid fragment was also part of a Pim-2 inhibitor previously identified through HTS by a group from Boehringer Ingelheim. The Bayer folks synthesized this molecule (compound 1), confirmed that it is also potent against Pim-1, and found that, crystallographically, it overlays beautifully with the fragment.
 The second paper, published in Bioorg. Med. Chem. Lett., describes how researchers at Genzyme used a fragment-based approach to develop potent Pim-1 inhibitors. The researchers used SPR to screen a library of about 1800 fragments at 75 micromolar concentration and then used a biochemical assay to characterize the active molecules. Benzofuran-2-carboxylic acids such as compound 10 (below) were particularly interesting: not only did they have high ligand efficiencies, they don’t look anything like typical kinase inhibitors. X-ray crystallography revealed that the bromine atom is binding in a hydrophobic pocket, while the acid is making hydrogen bond contacts with the catalytic lysine and other residues. A related fragment with comparable potency had a methoxy substituent in the 7-position, and by transforming this into a positively charged moiety the researchers were able to improve the potency to low nanomolar with a dramatic increase in ligand efficiency.
The second paper, published in Bioorg. Med. Chem. Lett., describes how researchers at Genzyme used a fragment-based approach to develop potent Pim-1 inhibitors. The researchers used SPR to screen a library of about 1800 fragments at 75 micromolar concentration and then used a biochemical assay to characterize the active molecules. Benzofuran-2-carboxylic acids such as compound 10 (below) were particularly interesting: not only did they have high ligand efficiencies, they don’t look anything like typical kinase inhibitors. X-ray crystallography revealed that the bromine atom is binding in a hydrophobic pocket, while the acid is making hydrogen bond contacts with the catalytic lysine and other residues. A related fragment with comparable potency had a methoxy substituent in the 7-position, and by transforming this into a positively charged moiety the researchers were able to improve the potency to low nanomolar with a dramatic increase in ligand efficiency.
 What’s also interesting is that if you overlay the two fragments from the two papers, they superimpose almost exactly on top of one another, with the carboxylic acid moieties making the same interactions – clearly a hot spot.
What’s also interesting is that if you overlay the two fragments from the two papers, they superimpose almost exactly on top of one another, with the carboxylic acid moieties making the same interactions – clearly a hot spot.
 On a side note, as most readers are probably aware, sanofi-aventis has recently acquired Genzyme. Practical Fragments wishes all the folks in Massachusetts well during the integration.
On a side note, as most readers are probably aware, sanofi-aventis has recently acquired Genzyme. Practical Fragments wishes all the folks in Massachusetts well during the integration.
In the first paper, published in Acta Cryst. Section D, researchers from Bayer Schering Pharma AG performed a crystallographic screen of just 36 fragments against Pim-1. Despite the small library size, a dozen of the fragments produced interpretable electron density, although only 4 of these showed activity better than 10 mM. Fragment 3, with an IC50 of 130 micromolar in an enzymatic assay, was the most potent. Interestingly, a close analog of this cinnamic acid fragment was also part of a Pim-2 inhibitor previously identified through HTS by a group from Boehringer Ingelheim. The Bayer folks synthesized this molecule (compound 1), confirmed that it is also potent against Pim-1, and found that, crystallographically, it overlays beautifully with the fragment.



16 May 2011
Native MS: turning up the voltage
At the FBDD meeting in San Diego last month there was some discussion of the connection between protein-ligand binding in the gas phase (ie, in native mass-spectrometry experiments) vs in solution; the discussion continued in the comment section here. Valerie Vivat, head of the mass spectrometry, molecular & cell biology department at NovAliX, has now provided a thorough response.
Valerie makes some interesting points here, in particular the statement that "the gas phase stability is not an indication of the complex binding affinity." Still, I do like the fact that native MS could be used as a quick way to sort enthalpic from entropic binders. What do you think?
When analyzing non-covalent complexes by mass spectrometry under non-denaturing conditions, one must keep in mind indeed how interactions stabilizing the complex in solution will be affected by the ion transfer in the gas phase. Basically, interaction based on hydrophobic effect is lost while the electrostatic-based interactions (Van der Waals, H-bonds, ionic interaction) are preserved (and are even strengthened due to the absence of solvent shielding). Regarding water molecules now, during the ESI process, biomolecules are transferred in the gas phase as partially hydrated ions and complete ion desolvation is subsequently achieved through low energy collisions with the residual gas molecules in the mass spectrometer interface. This complete desolvation is required for accurate mass measurements. Consequently, in most cases, molecular mass measurement of the complexes is accurate enough to rule out the possibility that water molecules remain after ions are transferred from the atmospheric pressure to the deep vacuum of the TOF analyzer, even in the case of crystallographic water molecules.
Regarding gas phase stability : Reports in the literature mention that collision-induced dissociation experiments and the determination of Vc50s can be used to monitor gain or loss in polar interaction. Indeed, we have now accumulated a variety of in house examples showing that increase in the gas phase stability of a complex is correlated with a gain in polar interactions as shown by X-ray or ITC. Monitoring the complex gas phase stability is thus an attractive feature to quickly evaluate, for example, gain or loss in polar interaction of analog series targeting the same protein binding site.
Importantly, the gas phase stability is not an indication of the complex binding affinity. Evaluation of complex affinity (after complexes are formed in solution) is done under instrumental conditions showing no dissociation of the non-covalent complexes. For complexes stabilized by electrostatic-based interactions (and thus rather stable in the gas phase), it is indeed much easier to find optimal instrument settings compared to complexes stabilized by hydrophobic effect. However, this does not preclude complexes mainly stabilized in solution by hydrophobic effect to be detected at all. For example, nuclear hormone receptors in complex with fatty acids or phospholipids are readily detected with appropriate settings. In this case, the polar head of the ligands is likely to act as a stable anchor in the gas phase, resulting in non-covalent complexes which remain intact during the ca. 1 millisecond flight of the ion from the ionization source to the detector.
Importantly, the gas phase stability is not an indication of the complex binding affinity. Evaluation of complex affinity (after complexes are formed in solution) is done under instrumental conditions showing no dissociation of the non-covalent complexes. For complexes stabilized by electrostatic-based interactions (and thus rather stable in the gas phase), it is indeed much easier to find optimal instrument settings compared to complexes stabilized by hydrophobic effect. However, this does not preclude complexes mainly stabilized in solution by hydrophobic effect to be detected at all. For example, nuclear hormone receptors in complex with fatty acids or phospholipids are readily detected with appropriate settings. In this case, the polar head of the ligands is likely to act as a stable anchor in the gas phase, resulting in non-covalent complexes which remain intact during the ca. 1 millisecond flight of the ion from the ionization source to the detector.
To sum up, native MS can be run under two different modes:
1. Low energy conditions where no complex dissociation occurs are used to assess the binding affinity of protein / ligand complexes,
2. High energy conditions inducing complex dissociation provide insight into the extent of polar interaction involved between the protein and the ligand. This type of experiment makes sense to compare a series of molecules binding to the same protein binding site.
Valerie makes some interesting points here, in particular the statement that "the gas phase stability is not an indication of the complex binding affinity." Still, I do like the fact that native MS could be used as a quick way to sort enthalpic from entropic binders. What do you think?
11 May 2011
Fragments vs ion channels
Ion channels represent an intriguing class of targets for a variety of indications, but it is difficult to develop robust high throughput assays. The gold standard is to actually measure the electrical conduction of the channels using techniques such as patch clamping, but this is time-consuming and tedious. Unfortunately, faster indirect methods are prone to high rates of false positives. Medium throughput automated patch clamp technologies are now available, and in a recent issue of Bioorg. Med. Chem. Lett. Scott Wolkenberg and colleagues at Merck decided to use one of these for a fragment screen.
The researchers screened 1280 fragments against the sodium ion channel acid-sensing ion channel 3 (ASIC3), a potential target to treat pain. Compounds were screened at 1 mM concentration, and 56 of them inhibited >50%. Interestingly, this 4.4% hit rate is comparable to other fragment screens at Merck on completely different target classes. Dose-response curves were generated for the 56 compounds, yielding 32 hits in 12 separate chemical series with ligand efficiencies > 0.3. Similarity searching led to the testing of 250 analogs, but most of these were less active than the original fragments.
In the absence of structural information, two strategies were used to try to improve the potencies. First, a series of 220 analogs were synthesized in which small (approximately 64 mass units) substituents were systematically added at various positions around the fragments. Unfortunately, most of the compounds were inactive, and those that were active were only marginally better than the starting fragment. The second strategy was to make more dramatic changes using parallel chemistry of up to a few hundred compounds. This was more successful: for example, fragment 3 led to compound 24, with 200-fold improved activity, albeit with a loss in ligand efficiency.
 Unfortunately the best compounds reported still do not break the micromolar barrier, and the pharmacokinetics are not encouraging. The authors conclude that:
Unfortunately the best compounds reported still do not break the micromolar barrier, and the pharmacokinetics are not encouraging. The authors conclude that:
The researchers screened 1280 fragments against the sodium ion channel acid-sensing ion channel 3 (ASIC3), a potential target to treat pain. Compounds were screened at 1 mM concentration, and 56 of them inhibited >50%. Interestingly, this 4.4% hit rate is comparable to other fragment screens at Merck on completely different target classes. Dose-response curves were generated for the 56 compounds, yielding 32 hits in 12 separate chemical series with ligand efficiencies > 0.3. Similarity searching led to the testing of 250 analogs, but most of these were less active than the original fragments.
In the absence of structural information, two strategies were used to try to improve the potencies. First, a series of 220 analogs were synthesized in which small (approximately 64 mass units) substituents were systematically added at various positions around the fragments. Unfortunately, most of the compounds were inactive, and those that were active were only marginally better than the starting fragment. The second strategy was to make more dramatic changes using parallel chemistry of up to a few hundred compounds. This was more successful: for example, fragment 3 led to compound 24, with 200-fold improved activity, albeit with a loss in ligand efficiency.

The present results are consistent with the analysis that advancing fragment hits to high potency (<100 nM) and/or to a preclinical development milestone in the absence of structural information is unlikely.I think this may be overly pessimistic. While optimizing fragments in the absence of structure is certainly challenging, it is possible. Fragment screening ultimately delivered several distinct series of low micromolar compounds against ASIC3. Had these come from an HTS assay, the job of optimizing them further would not be any easier. Moreover, this report represents another example of using fragment approaches to tackle membrane proteins.
29 April 2011
Not fragments versus DOS, fragments from DOS
A few months ago we highlighted a forum in Nature comparing fragment-based lead discovery with diversity-oriented synthesis, or DOS. This was quite a vigorous debate and was covered on our sister blog as well as In the Pipeline and second messenger. Personally I’ve never been a this or that kind of guy – more of a this and that – so it is refreshing to see a paper in this week’s issue of PNAS describing a DOS approach to building fragments.
Damian Young and colleagues at Harvard and the Broad Institute, ground zero for DOS, noted that many commercial fragments contain a sizable percentage of sp2 carbons: aromatic rings, for example. Because a larger number of aromatic rings correlates with lower solubility and higher attrition in lead development, the researchers focused on using DOS to generate fragments that would have a higher fraction of sp3 carbons at the expense of sp2 carbons. They used a “build/couple/pair” approach, in which chiral “building blocks” (in this case proline derivatives) were “coupled” to another building block and then functional groups were “paired” to generate bicyclic molecules. The result was about three dozen fragments.
So how do they look? Actually, not so bad. Superficially they all resemble one another, but because they contain up to three stereocenters they cover quite a bit of chemical space while still conforming to the rule of 3. Significantly, they are in fact more three-dimensional than commercially available fragments (from ZINC) having the same molecular formula or the same set of calculated physical properties (molecular weight, cLogP, number of hydrogen-bond donors and acceptors, etc.). The DOS fragments contain a larger fraction of methyl esters and carboxylic acids than I would want to see in a library overall, but this was intentional, and none of them are downright ugly.
Unfortunately the paper provides no screening data, so it is anyone’s guess whether any of the fragments will turn out to be active. Still, the approach is likely to probe new areas of chemical space. Hopefully some of the commercial purveyors of fragments will start making and selling these types of molecules.
Damian Young and colleagues at Harvard and the Broad Institute, ground zero for DOS, noted that many commercial fragments contain a sizable percentage of sp2 carbons: aromatic rings, for example. Because a larger number of aromatic rings correlates with lower solubility and higher attrition in lead development, the researchers focused on using DOS to generate fragments that would have a higher fraction of sp3 carbons at the expense of sp2 carbons. They used a “build/couple/pair” approach, in which chiral “building blocks” (in this case proline derivatives) were “coupled” to another building block and then functional groups were “paired” to generate bicyclic molecules. The result was about three dozen fragments.
So how do they look? Actually, not so bad. Superficially they all resemble one another, but because they contain up to three stereocenters they cover quite a bit of chemical space while still conforming to the rule of 3. Significantly, they are in fact more three-dimensional than commercially available fragments (from ZINC) having the same molecular formula or the same set of calculated physical properties (molecular weight, cLogP, number of hydrogen-bond donors and acceptors, etc.). The DOS fragments contain a larger fraction of methyl esters and carboxylic acids than I would want to see in a library overall, but this was intentional, and none of them are downright ugly.
Unfortunately the paper provides no screening data, so it is anyone’s guess whether any of the fragments will turn out to be active. Still, the approach is likely to probe new areas of chemical space. Hopefully some of the commercial purveyors of fragments will start making and selling these types of molecules.
23 April 2011
Ligandability
The sequencing of the human genome has thrown up lots of potential targets, but choosing which ones to pursue is difficult: many are not biologically relevant and many are shaped such that small molecules are unable to affect their activity. “Druggability” is a popular neologism that captures both of these ideas; it refers to whether a protein can be targeted by a small molecule – preferably an orally bioavailable one – to treat a disease. However, the two components of druggability are really separate concepts, and in this month’s issue of Drug Discovery Today Fredrik Edfeldt, Rutger Folmer, and Alex Breeze coin a new term – “ligandability”. A protein is ligandable if potent small-molecule ligands can be found for it. Obviously for a protein to be druggable it needs to be ligandable, and thus it would be nice to assess this characteristic as quickly as possible. How can this be done?
Enter fragments. Because fragments have lower complexity than lead-sized (let alone drug-sized) molecules, hit rates from fragment screens tend to be higher. If a binding pocket exists in a protein, a small library of just 1000 fragments or so should produce a good range of hits. In fact, Phil Hajduk and colleagues at Abbott found several years ago that fragment screens predict the success of lead discovery campaigns. In the new paper, Edfeldt and colleagues, all at AstraZeneca, analyzed 36 internal discovery projects where both fragment screens and HTS had been conducted. They used data from the fragment screens to categorize targets into three ligandability bins:
AstraZeneca is now using fragment-based ligandability screening to help assess which targets to pursue: those with low ligandability are only pursued when the biology is truly compelling. On the flip side, targets that have failed conventional HTS but have high ligandability are reexamined using alternative hit discovery techniques, such as fragment-based methods. This seems like an appealing approach: fragments not only help drug hunters avoid throwing out the baby with the bathwater, but also to avoid drowning in dirty bathwater. I wonder how many other companies are using similar strategies.
Enter fragments. Because fragments have lower complexity than lead-sized (let alone drug-sized) molecules, hit rates from fragment screens tend to be higher. If a binding pocket exists in a protein, a small library of just 1000 fragments or so should produce a good range of hits. In fact, Phil Hajduk and colleagues at Abbott found several years ago that fragment screens predict the success of lead discovery campaigns. In the new paper, Edfeldt and colleagues, all at AstraZeneca, analyzed 36 internal discovery projects where both fragment screens and HTS had been conducted. They used data from the fragment screens to categorize targets into three ligandability bins:
- Low: low hit rate, best affinities > 1 mM, low diversity of hits
- Medium: intermediate hit rate, best affinities 0.1 – 1 mM, some diversity of hits
- High: high hit rate, best affinities < 0.1 mM, high diversity of hits
AstraZeneca is now using fragment-based ligandability screening to help assess which targets to pursue: those with low ligandability are only pursued when the biology is truly compelling. On the flip side, targets that have failed conventional HTS but have high ligandability are reexamined using alternative hit discovery techniques, such as fragment-based methods. This seems like an appealing approach: fragments not only help drug hunters avoid throwing out the baby with the bathwater, but also to avoid drowning in dirty bathwater. I wonder how many other companies are using similar strategies.
15 April 2011
Sixth Annual Fragment-Based Drug Discovery
The only US-based conference completely devoted to fragment-based drug discovery ended in San Diego this week. As with last year, I won’t attempt to summarize all of the talks – there was far more information presented than I have time to write (or that you probably have patience to read!) For those of you who were there, please feel free to mention some of the things I missed.
One of the points that Don Huddler (GlaxoSmithKline) and I (Carmot) made in the pre-conference short-course is that finding fragments is a solved problem. As Rod Hubbard (Vernalis, University of York) noted in his opening presentation, “it’s pretty simple to find fragments that bind; a graduate student can do it in a couple months.” Even membrane proteins are starting to yield to fragment-based screening, as Gregg Siegal (ZoBio, Leiden University) discussed in his closing session (see also here).
That’s not to say that new methods for finding fragments aren’t useful, particularly if they open new target space, are faster or more reliable, or provide new information. An example of the latter was the presentation by Denis Zeyer (NovAliX) on native mass-spectrometry (see also here). Because hydrophobic interactions are weaker in the gas phase than in water, it should be possible to select for molecules that bind predominantly through polar interactions. In fact, by gradually increasing the voltage in their MS instrument, Zeyer and colleagues generated “VC50” curves, the voltage at which half the compound dissociates from the protein. At least in one case, a higher VC50 correlated with the presence of an additional hydrogen bond to the protein compared with related molecules.
Polar contacts are generally associated with enthalpic rather than entropic interactions, and whether such fragments are preferable was the subject of some discussion, particularly at a breakfast round-table discussion. In contrast to a meeting just last year, several participants were actively collecting thermodynamic data, though there was some uncertainty as to what to do with it. This is a controversial subject; one person suggested that enthalpic binders are likely to be more hydrophilic than entropic binders, so just keeping an eye on lipophilicity is likely to be just as useful and far easier than actually measuring thermodynamic parameters. Charles Reynolds (Ansaris) provided an analysis that illustrates some of the difficulties in using thermodynamic data – I’ll follow up on this in a later post.
The shape of fragments has been previously discussed, and Ivan Efremov (Pfizer) gave a nice case study of a strikingly three-dimensional fragment: an X-ray screen of 340 molecules against BACE resulted in a single hit, a spirocyclic pyrrolidine. The electron density of this was so clear that it didn’t even need to be deconvoluted from the other three compounds in the pool, and medicinal chemistry ultimately led to low micromolar inhibitors.
There was general consensus that ligand efficiency (and various lipophilicity adjusted versions) is a helpful metric. One practitioner said that his company had sometimes pursued more chemically tractable but less ligand efficient fragments and generally came to regret those decisions. But a fragment with lower ligand efficiency could still be interesting: with fragments, even small changes could have huge effects on binding (see for example AT13387, which was discussed by Chris Murray of Astex). Thus, a bit of initial fragment optimization could be a good investment before pursuing more intensive chemistry, particularly if commercial or in-house analogs are available. Interestingly, I couldn’t find anyone who uses either fit quality or %LE.
In the early days of fragment-based lead discovery a common selling point was that it sped up drug discovery, but a theme in this meeting was that it is not necessarily faster but can provide leads against more difficult targets or better leads against “normal” targets. Of course, one has to be wary of taking a good fragment, slapping a bunch of grease on it, and turning it into a lipophilic monster.
Indeed, an analysis of fragment-derived leads published a couple years ago was not flattering. Taking up the thrown gauntlet on behalf of fragments, Chris Murray presented a retrospective analysis of all 42 fragment to lead programs at Astex (including 21 kinases and 9 proteases). The average parameters of these leads were considerably more attractive in terms of both molecular weight and ClogP that the published values of the HTS hits. At least according to this analysis, fragment approaches have the potential to deliver superior molecules, as long as one is disciplined and creative in how these approaches are applied.
One of the points that Don Huddler (GlaxoSmithKline) and I (Carmot) made in the pre-conference short-course is that finding fragments is a solved problem. As Rod Hubbard (Vernalis, University of York) noted in his opening presentation, “it’s pretty simple to find fragments that bind; a graduate student can do it in a couple months.” Even membrane proteins are starting to yield to fragment-based screening, as Gregg Siegal (ZoBio, Leiden University) discussed in his closing session (see also here).
That’s not to say that new methods for finding fragments aren’t useful, particularly if they open new target space, are faster or more reliable, or provide new information. An example of the latter was the presentation by Denis Zeyer (NovAliX) on native mass-spectrometry (see also here). Because hydrophobic interactions are weaker in the gas phase than in water, it should be possible to select for molecules that bind predominantly through polar interactions. In fact, by gradually increasing the voltage in their MS instrument, Zeyer and colleagues generated “VC50” curves, the voltage at which half the compound dissociates from the protein. At least in one case, a higher VC50 correlated with the presence of an additional hydrogen bond to the protein compared with related molecules.
Polar contacts are generally associated with enthalpic rather than entropic interactions, and whether such fragments are preferable was the subject of some discussion, particularly at a breakfast round-table discussion. In contrast to a meeting just last year, several participants were actively collecting thermodynamic data, though there was some uncertainty as to what to do with it. This is a controversial subject; one person suggested that enthalpic binders are likely to be more hydrophilic than entropic binders, so just keeping an eye on lipophilicity is likely to be just as useful and far easier than actually measuring thermodynamic parameters. Charles Reynolds (Ansaris) provided an analysis that illustrates some of the difficulties in using thermodynamic data – I’ll follow up on this in a later post.
The shape of fragments has been previously discussed, and Ivan Efremov (Pfizer) gave a nice case study of a strikingly three-dimensional fragment: an X-ray screen of 340 molecules against BACE resulted in a single hit, a spirocyclic pyrrolidine. The electron density of this was so clear that it didn’t even need to be deconvoluted from the other three compounds in the pool, and medicinal chemistry ultimately led to low micromolar inhibitors.
There was general consensus that ligand efficiency (and various lipophilicity adjusted versions) is a helpful metric. One practitioner said that his company had sometimes pursued more chemically tractable but less ligand efficient fragments and generally came to regret those decisions. But a fragment with lower ligand efficiency could still be interesting: with fragments, even small changes could have huge effects on binding (see for example AT13387, which was discussed by Chris Murray of Astex). Thus, a bit of initial fragment optimization could be a good investment before pursuing more intensive chemistry, particularly if commercial or in-house analogs are available. Interestingly, I couldn’t find anyone who uses either fit quality or %LE.
In the early days of fragment-based lead discovery a common selling point was that it sped up drug discovery, but a theme in this meeting was that it is not necessarily faster but can provide leads against more difficult targets or better leads against “normal” targets. Of course, one has to be wary of taking a good fragment, slapping a bunch of grease on it, and turning it into a lipophilic monster.
Indeed, an analysis of fragment-derived leads published a couple years ago was not flattering. Taking up the thrown gauntlet on behalf of fragments, Chris Murray presented a retrospective analysis of all 42 fragment to lead programs at Astex (including 21 kinases and 9 proteases). The average parameters of these leads were considerably more attractive in terms of both molecular weight and ClogP that the published values of the HTS hits. At least according to this analysis, fragment approaches have the potential to deliver superior molecules, as long as one is disciplined and creative in how these approaches are applied.
07 April 2011
Wedding announcement: SuperGen and Astex
It’s wedding season in the world of fragments – last month we noted the merger of Daiichi Sankyo and Plexxikon, and today SuperGen and Astex Therapeutics announced that they are tying the knot. Unfortunately the “dowry” in this case is considerably smaller: SuperGen will pay Astex shareholders $25 million cash upfront, with an additional $30 million in deferred payments. Astex shareholders will, however, still retain 35% equity in the combined company.
Astex has long been a leader in fragment-based drug discovery, having taken at least three programs from fragments to clinical compounds (see blog entries on AT13387, AT9283, and AT7519). Perhaps it is in recognition of this that the combined entity will be called Astex Pharmaceuticals. Practical Fragments wishes everyone involved the best of luck.
Astex has long been a leader in fragment-based drug discovery, having taken at least three programs from fragments to clinical compounds (see blog entries on AT13387, AT9283, and AT7519). Perhaps it is in recognition of this that the combined entity will be called Astex Pharmaceuticals. Practical Fragments wishes everyone involved the best of luck.
01 April 2011
Fragments in vivo
The number of ways to find fragments just keeps growing. A few weeks ago we discussed WAC, which takes its place alongside ITC, SPR, MS, TINS, and more traditional methods such as NMR, X-ray and biochemical screening. However, all of these approaches are somewhat reductionist, relying on isolated target proteins. In an effort to bring the whole organism into the picture, our friends at the University of Shutka, Russia, have come up with an approach they call “Fragments in Bodies,” or FIB.
The researchers have assembled a collection of very small fragments, purchased for the most part from Lilliput Pharmaceuticals. These are then screened in mouse models to look for positive phenotypic effects.
The researchers face some unique challenges. For example, it is difficult to measure changes in body mass as the animals need to consume such large amounts of fragments that they can become somewhat bloated. Still, if the animals can be safely dosed with massive amounts of micro-molecules, "FIB"ing could provide very good starting points for further work!
The researchers have assembled a collection of very small fragments, purchased for the most part from Lilliput Pharmaceuticals. These are then screened in mouse models to look for positive phenotypic effects.
The researchers face some unique challenges. For example, it is difficult to measure changes in body mass as the animals need to consume such large amounts of fragments that they can become somewhat bloated. Still, if the animals can be safely dosed with massive amounts of micro-molecules, "FIB"ing could provide very good starting points for further work!
Subscribe to:
Posts (Atom)

