The research group had previously conducted a fragment screen against Hsp90, resulting in a number of hits. In the new paper, fragment hits 1 and 2 (see figure) were both found to have fairly low affinities, but were characterized crystallographically. Interestingly, fragment 2 could adopt at least two very different conformations, depending on whether it was co-crystallized in the presence of fragment 1. In the ternary structure, the two fragments come within about 3 Å of each other, and molecular modeling suggested that four atoms should be able to link them.
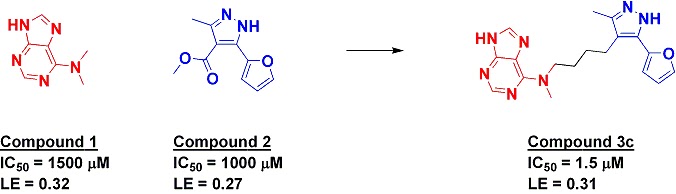
Gratifyingly, when such a compound was made and tested, it inhibited the enzyme several hundred-fold more tightly than either of the initial fragments. The crystal structure revealed that the compound binds similarly to the ternary structure of Hsp90 and fragments 1 and 2.
The authors note that “the binding free energy of the linked fragment 3c was found to be exactly the sum of those of the original two fragments.” Of course, this is still a long way from an ideal linking situation: as noted earlier this year a good linker should lead to super-additivity (an improvement of ligand efficiency), not just additivity (maintenance of ligand efficiency). Nonetheless, this example is still better than many attempts at linking, which often are less than additive.


No comments:
Post a Comment