Proteins in cells are constantly synthesized and
degraded in a complex, highly regulated manner managed in part by the ubiquitin
proteasome system. Simplistically, a ubiquitous small protein called ubiquitin
is conjugated to other proteins, targeting them for destruction, and some of
the proteins thus targeted control the stability of still other proteins. But ubiquitination
is not destiny: ubiquitin can be removed by more than 100 deubiquitinating
enzymes, or DUBs.
As I said, this is complex. But complexity has never stopped
folks from pursuing drug targets, and multiple groups are interested in a
particular DUB called USP7, which is implicated in cancer and other
indications. USP7 is one of more than 50 members of a subfamily of DUBs that use cysteine as a catalytic residue. Selectivity is an obvious challenge, and
since cysteine is chemically reactive, any screening result carries a high risk
of being an artifact. Two recent papers describe how fragment-based approaches
led to potent, selective inhibitors.
The first, published in J.
Med. Chem. by Paola Di Lello, Vicki Tsui, and coworkers at Genentech,
started with an NMR fragment screen. This identified molecules such as compound
1, which NMR data suggested bound near the active-site cysteine. This and other
fragments were used to conduct virtual screens of the much larger Genentech
library, and 21 of these were then tested experimentally. Most of these either
didn’t bind, bound to multiple sites, or caused protein aggregation, but four
of them, including compound 2, showed clear binding to a specific site on USP7
and also inhibited the enzyme in a biochemical assay.
Surprisingly, protein-detected NMR suggested that these four
molecules did not bind in the active site as expected but rather in an adjacent
“palm site”, a hypothesis that was confirmed by a crystal structure of compound
2 bound to USP7. This led the researchers to reexamine other hits from the
original NMR screen, where they identified several aminopyridinephenols, such
as compound 13.
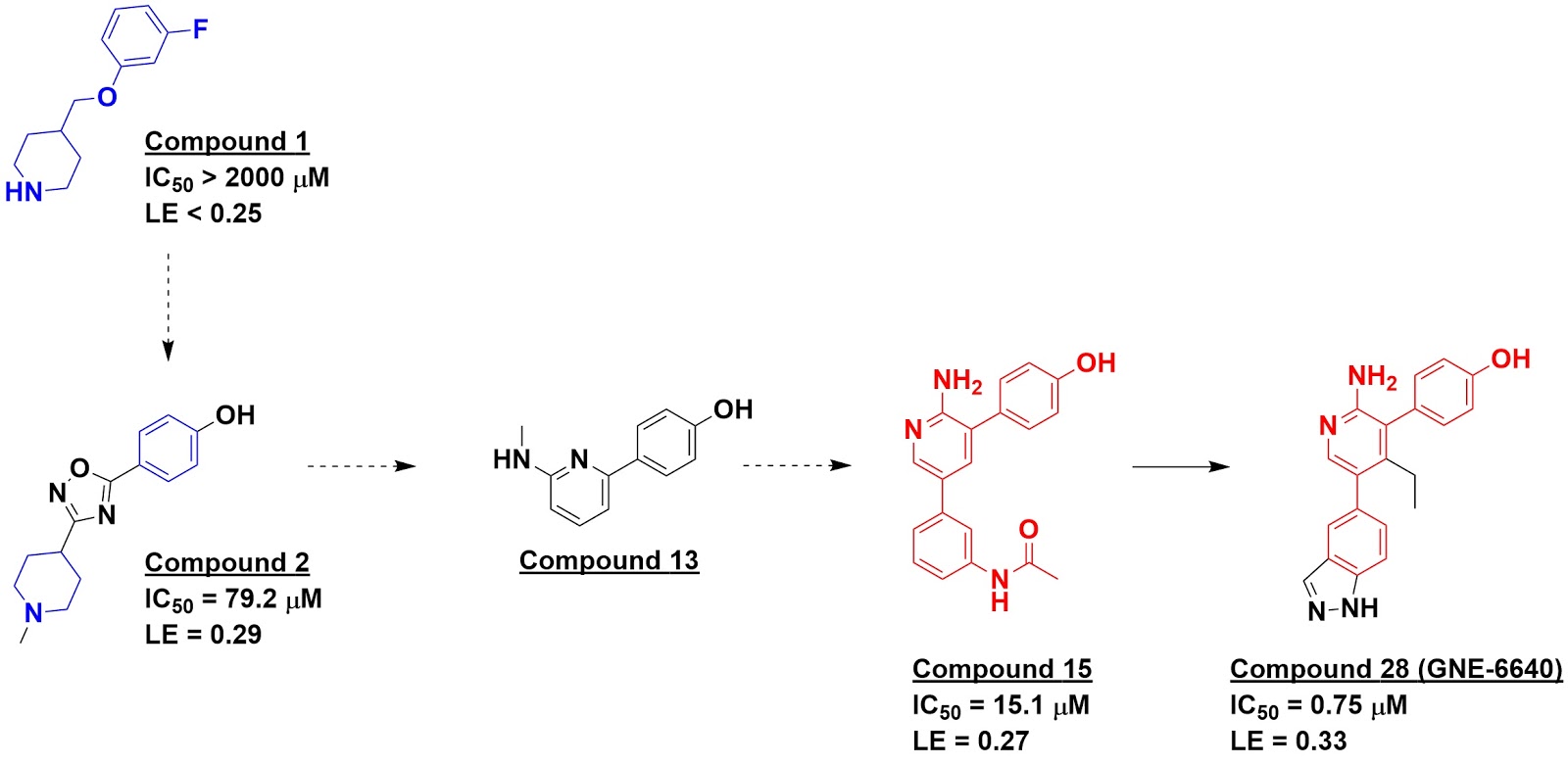
Meanwhile, a biochemical HTS against USP7 had identified 76 hits, but most of these turned out to be artifacts, and none of them yielded co-crystal structures with the enzyme. The fragment findings led the researchers to revisit some of the weaker hits that had been overlooked, such as compound 15. This led to a crystal structure showing binding in the palm site, and further medicinal chemistry ultimately led to molecules such as compound 28 (GNE-6640), with nanomolar activity in both biochemical and cell-based assays. A separate paper in Nature characterizes the biology in more detail, revealing that molecules in this series interfere with ubiquitin binding and are highly selective for USP7.
Another fragment effort on this target was reported by Timothy
Harrison and collaborators at Almac and Queen’s University, Belfast in Nat. Chem. Biol. An SPR screen of 1946
fragments against the catalytic domain of USP7 led to compounds such as
fragment B. This was combined with molecules from other groups that had been
reported in the literature, leading to compound 1. Subsequent medicinal
chemistry, informed by crystallography, led to compound 4, with low nanomolar
biochemical and cell-based activity and excellent selectivity. The enantiomer
is much less active, and compound 4 should be a useful chemical probe to
further understand the biology of USP7.


Remarkably, not only do the two series of molecules bind
some distance away from the active site cysteine (yellow, upper right), they bind in completely different, non-overlapping sites!
These papers illustrate the importance of allosteric sites for tackling specific members of large protein families. They are also both
cases of “fragment-assisted drug discovery.” Unlike many success stories we’ve
highlighted, it is difficult or impossible to find the initial fragment in the
final molecules. Heck, Genentech’s best molecules bind in a completely
different site from where the first fragment hits bound. Being open to such
possibilities, and using all available data from every possible source, are keys
to success.


4 comments:
In my experience, gained from several FBDD screens, many proteins containing catalytic cysteines have proven to be 'challenging' or 'refractory' to FBDD. Such screens are frequently associated with conformational change and aggregation during fragment screening or following cysteine oxidation, making it difficult to separate useful hits from 'false positives' and to pursue hits using standard SBDD methods.
My suspicion is that cells have developed mechanisms to recognise when catalytic cysteines have become irreversibly oxidised [oxidation state S(+2) and above] and that conformation change and aggregation are part of these mechanisms. The proteins may exist on a conformational 'knife edge' and so are sensitive to small changes around the cysteine. In rare cases, protective mechanisms exist which can reversibly trap a lower oxidation state [S(0)] - the sulphenyl amide of PTP1B is an example. However in the remaining cases, fragments that can catalyse oxidation, for example even very weak redox-cyclers, and many that simply bind, may find the site 'booby-trapped' and lead to the triggering of adverse changes.
Hi Glyn,
Thanks, this is a really interesting hypothesis! I wonder if it would be possible to test using a cellular assay such as CETSA, comparing for example cysteine hydrolases with serine hydrolases to see if the former are indeed more sensitive to stress.
Hi Dan,
In a cellular context, I guess that you would want to look at the effects of mild oxidative stress on protein turnover, but I am not sure how you would define 'mild' or how you would measure turnover under these conditions.
For practical FBDD applications, I would consider 'stress testing' protein targets that contain active cysteines before screening, using any known (or suspected) redox cyclers and 'frequent hitters' in the library e.g. quinols. That should give a sense of the sensitivity of the target. If the 'booby trap' is sprung by sulphur oxidation, then the conformational trigger may be the presence of a strong acid anion in, or near, the site. It would be interesting to know if small anionic fragments form a class of 'false positives' for these targets.
I would also like to believe that the reversible formation of sulphenyl amides to protect the sulphur(+2) oxidation state is more common that has been noted. The expected mass change (-2Da) is detectable in some cases and the modification has been observed in X-ray structures, but these are not generally accessible experiments.
A series of DUBs have been assessed for sensitivity to oxidation and the authors hypothesize this sensitivity is tied to novel regulatory mechanisms in vivo.
http://www.nature.com/articles/ncomms2532
Interestingly, USP7 is not as sensitive to oxidation as other DUBs, possibly due to conformational changes in the apo form of the enzyme that these allosteric inhibitors target.
Post a Comment