Machine learning has become a hot
new thang in drug discovery, attracting massive attention and investment. While
easy to parody, artificial intelligence techniques are becoming increasingly
powerful. A new paper in J. Chem Inf. Mod. by Angelo Pugliese and colleagues at
the Beatson Institute applies the methodology to generate a new fragment library.
Machine learning entails collecting
large amounts of data, passing that through various neural networks, and obtaining
recommendations. In this case, the researchers wanted to generate “privileged fragments” that would hit in multiple assays. (Of course, the idea would be to
make genuinely privileged fragments, such as 4-azaindole, rather than PAINS.)
The researchers used a training set of 66 fragments that hit in at least three
of 25 screens done at the Beatson, for which the average hit rate was
2.18%.
First though, the researchers
needed to teach their model how to generate chemically valid fragments in the
first place (for example, fewer than 5 bonds to carbon). To do this they used
both SMILES (simplified molecular-input line-entry system) and chemical
fingerprints from a set of 486,565 commercially available fragments. They then
combined this model with the privileged fragments. Extensive details are provided;
as they go well beyond my expertise I won’t even attempt to summarize them. (For
example, “the classifier for the smi2smi model comprised sequential 64-unit and
32-unit dense ReLU layers followed by a single sigmoid output neuron.”) At the
end of the exercise, and after triaging by medicinal chemists, the researchers
came up with a set of 741 fragments.
What are their overall properties? For
one thing, generated fragments tend to be more planar (as assessed by PBF) and
have lower Fsp3 values than the nearly half-million fragments used
for training. The researchers acknowledge that this could reflect the historical
composition of the Beatson fragment library, although as we argued here it
could also be true that flatter fragments just give higher hit rates.
Molecular complexity is a fundamental
but poorly defined aspect of fragment-based lead discovery, and the researchers
have come up with their own metric, called feature complexity (FeCo), which
incorporates information on rotatable bonds, numbers of halogens, hydrogen bond
donors and acceptors, charged groups, aromatic rings, and hydrophobic elements,
all normalized by the number of heavy atoms. Hopefully this will be explored
more fully in a dedicated publication.
What do the individual fragments
actually look like? Five examples are shown in the paper, and nearly 200 more
are provided in the supporting information. Below are seven chosen arbitrarily from
that list (sampling every 30 structures).
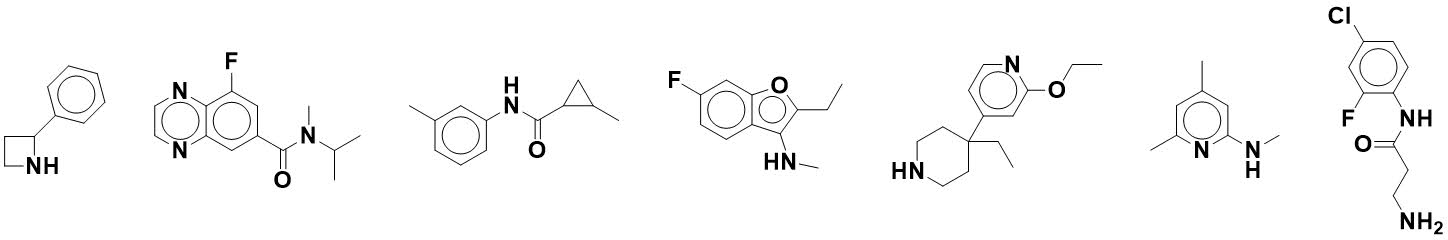
Of course, the question remains as
to whether these fragments will truly turn out to be privileged. As might be
expected given the vastness of chemical space, only 78 of the 741 are
commercially available. The researchers note that they are acquiring some of
these, and it will be interesting to see how they perform in the screens to
come.


No comments:
Post a Comment