To find inhibitors against PDK1, we used Tethering with extenders. This approach starts by covalently modifying a protein of interest with an “extender,” which is a fragment designed to bind in a desired site on a protein and to capture other fragments; it contains a protein-reactive functionality as well as a masked thiol. In this case, we used a generic fragment known to interact with the purine-binding pocket of kinases, a diaminopyrimidine (red in upper left of figure). The protein-extender complex was then screened against a library of several thousand disulfide-containing fragments under partially reducing conditions; only fragments with some affinity for the protein will remain covalently bound to the protein and thus be detectable by mass-spectrometry. The pyridinone (blue in figure) was one of the strongest hits.
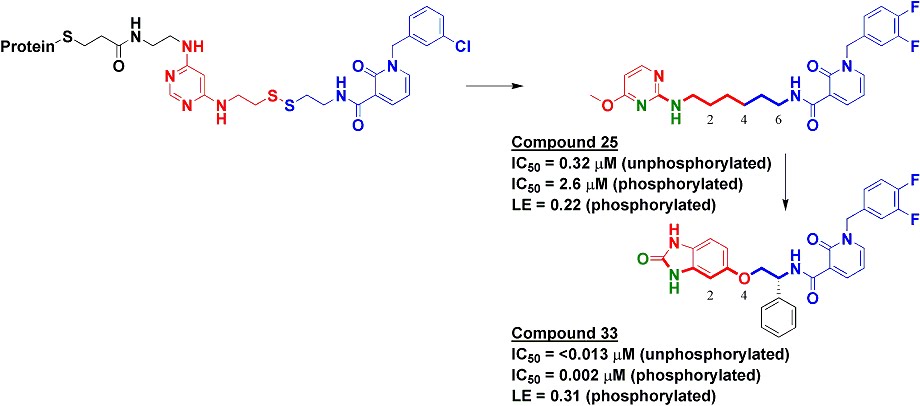
We synthesized several molecules containing purine-pocket binding elements connected to pyridinones by flexible linkers and found compound 25 to be one of the most potent. The SAR revealed the importance of a hydrogen-bond donor-acceptor pair (green atoms in figure) a specific distance from the pyridinone fragment, and further medicinal chemistry led ultimately to compound 33. In addition to being quite potent, compound 33 was remarkably selective for PDK1: in a panel of 241 kinases screened at 10 micromolar concentration of compound 33, only PDK1 and one other kinase were inhibited by 80% or more.
The selectivity of compound 33 was partially explained by the X-ray crystallographic structure, which revealed that the pyridinone fragment discovered through Tethering binds in the so-called adaptive pocket with the activation loop in the “DFG-out” conformation. I believe this is the only series of inhibitors for which this binding mode has been observed for PDK1. Consistent with the structure, these inhibitors are more potent against the unphosphorylated (inactive) form of PDK1 than against the phosphorylated (active) form.
Compound 33 was effective at preventing phosphorylation of the PDK1 substrate Akt both in cells as well as in a xenograft model. Though I may be biased in thinking so, this is a nice application of a fragment-based approach to discover a strikingly specific kinase inhibitor. I hope that people will use compound 33 to further probe the biology of the PI3K pathway. Indeed, at least one completely independent team of researchers seems to already be doing so.


No comments:
Post a Comment