Advancing fragments without high-resolution structural information
remains a challenge scientists often choose not to take on, according to our
poll last year. But for many appealing targets, such as membrane proteins,
structural information is difficult to obtain. In a new paper in J. Med. Chem., Peter Kolb and
collaborators at Philipps-University Marburg and Vrije Universiteit Brussel
describe a computational strategy.
The approach, called “growing via merging”, starts with a
core fragment that binds to a target, in this case the β2-adrenergic receptor (β2AR). Ideally this interaction is structurally
characterized, but if not a model can suffice. Here, the researchers started with
five fragments they had previously discovered. All of these had in common a
lipophilic core with a primary or secondary amine appendage; this is a known
pharmacophore for β2AR, so
modeling could be used to orient the fragments.
Next, this core fragment is derivatized in silico with other
fragments using a selection of 58 common reactions. Since all five fragments
contained an amine, reductive amination was used here. A set of nearly 19,000
fragment-sized aldehydes and ketones was extracted from the ZINC database and computationally
transformed into amines – as if they were reacted with one of the core
fragments. These were then docked into the receptor, and those that did not
overlap with the core fragments and also placed the amine near the amine of the
core fragment were kept for further analysis.
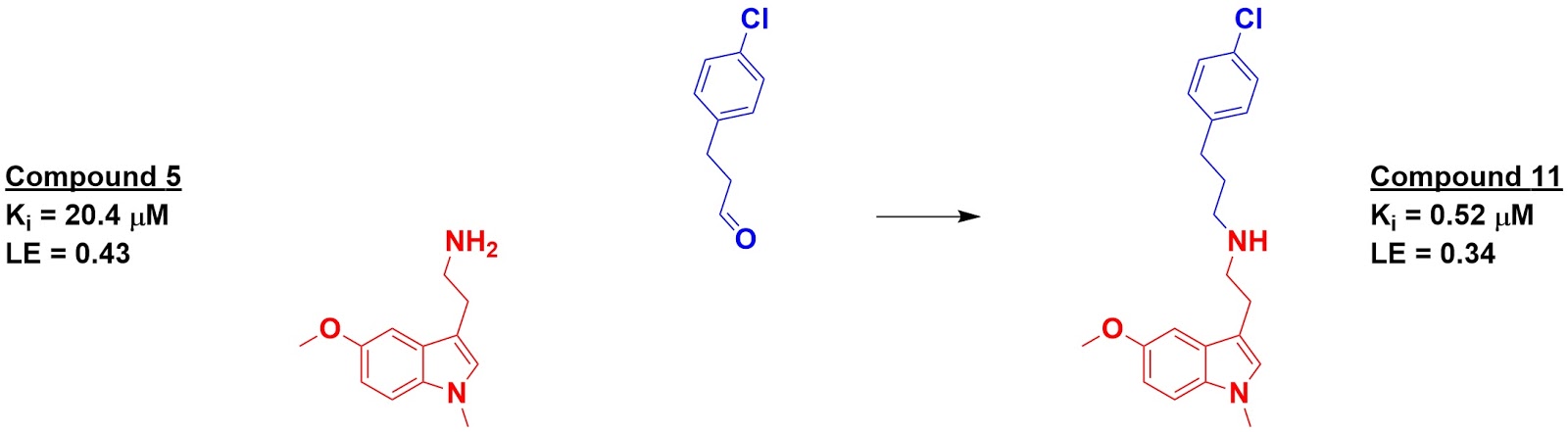
The top 500-scoring fragments were then “reacted” – again in
silico – with the core fragments and again docked. Eight of these were actually
synthesized and tested for binding, of which four had higher affinity than the initial
fragments. The best, compound 11, showed a 40-fold boost in affinity over its
starting fragment.