The researchers started by assembling a small (113 compound)
fragment library designed to target aspartic proteases. This was screened
against renin by NMR, resulting in hits such as compound 3. Although these were
too weak to yield dissociation constants or IC50 values by NMR or
biochemical screens, the researchers were able to obtain crystal structures of
at least two of these fragments bound to renin, including compound 3.
Interestingly, although the amino alcohol moiety of this compound was designed
to target the catalytic aspartic acids, this turned out not to be the case. Instead the
binding appears to be largely driven by hydrophobic contacts between the
tricyclic moiety of the fragment and the so-called S3-S1 pocket
of the protein.
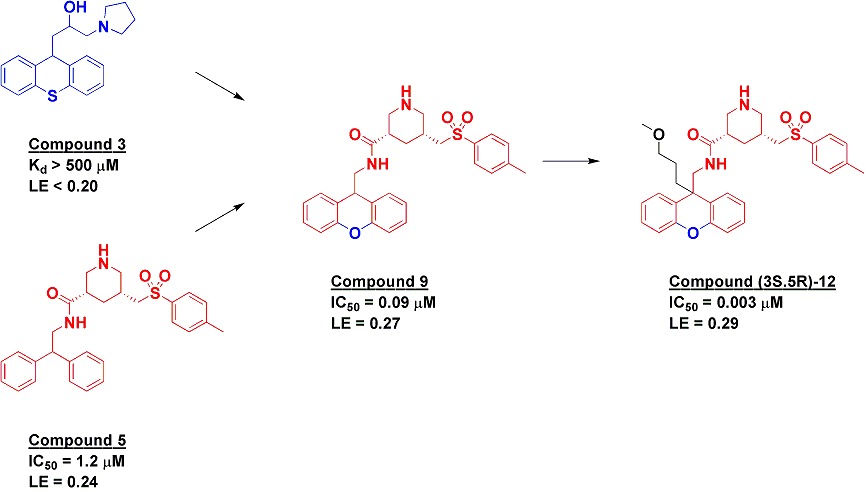
Along with the fragment effort, the researchers also
undertook an HTS screen, resulting in the discovery of compound 5, which itself
had come from a 950-compound library targeted towards aspartic proteases.
Crystallography revealed that this molecule binds with the diphenylmethane
moiety in a similar position as the tricycle of fragment 3, and indeed when a
rigid tricyclic framework was grafted onto compound 5 the resulting compound 9
showed a satisfying boost in potency. Further optimization led to a pure
enantiomer of compound 12 with low nanomolar potency, good selectivity,
moderate oral bioavailability and efficacy in rats, though it did also show
some time-dependent CYP3A4 inhibition.
This is really a structure-based design paper, and there is
obviously much more detail than would be appropriate here. What caught my eye
is that it is a nice example of fragment-assisted drug discovery, in which
fragment information is used as one aspect of an overall lead discovery
program. In this case a cynic could argue that the only bit that came from the
fragment was the tricylic motif. However, given the limitless number of analogs
that could be made, such information can be both unexpected and valuable.


No comments:
Post a Comment