One of the cornerstones underpinning fragment-based lead
discovery is molecular complexity: fragments are less complex than larger
molecules, and are thus likely to bind to more sites on more proteins. In
theory, then, you want relatively simple fragments, and in fact Astex has
actually formalized this with the concept of the “minimal pharmacophore”, in
which each fragment contains a single pharmacophore (such as a hydrogen bond
donor next to a hydrogen bond acceptor). But this is not the only way to build
a fragment library; in 2016 we noted a paper out of the University of Dundee
describing fragment libraries built with “caps” for easy derivatization. In a
new paper in ChemMedChem, Paul Wyatt,
Peter Ray, and collaborators at the University of Dundee and GlaxoSmithKline describe
a screen with this “functional group complexity” (FGC) library.
The researchers were interested in the protein InhA, a drug
target for Mycobacterium tuberculosis,
the organism causing the eponymous disease. A relatively small library of 1360
fragments was assembled from six different sources, loosely defined by the authors:
- 573 commercial fragments
- 170 “3D” fragments from the 3DFrag consortium
- 326 of the designed FGC fragments
- 46 commercial fragments chosen based on known InhA inhibitors
- 124 “inventory” fragments
- 121 “project” fragments
Previous work had suggested that more potent molecules
seemed to reduce the STD signals for the NADH cofactor, so these molecules (32
fragments) were prioritized. The 13 FGC fragments represented a hit rate of 4%,
nearly double the 2.4% for the library as a whole.
All 149 of the initial fragments were tested in a
biochemical assay at 0.5 mM, but only 4 gave measurable inhibition – too few to
draw conclusions. Five compounds were characterized crystallographically bound
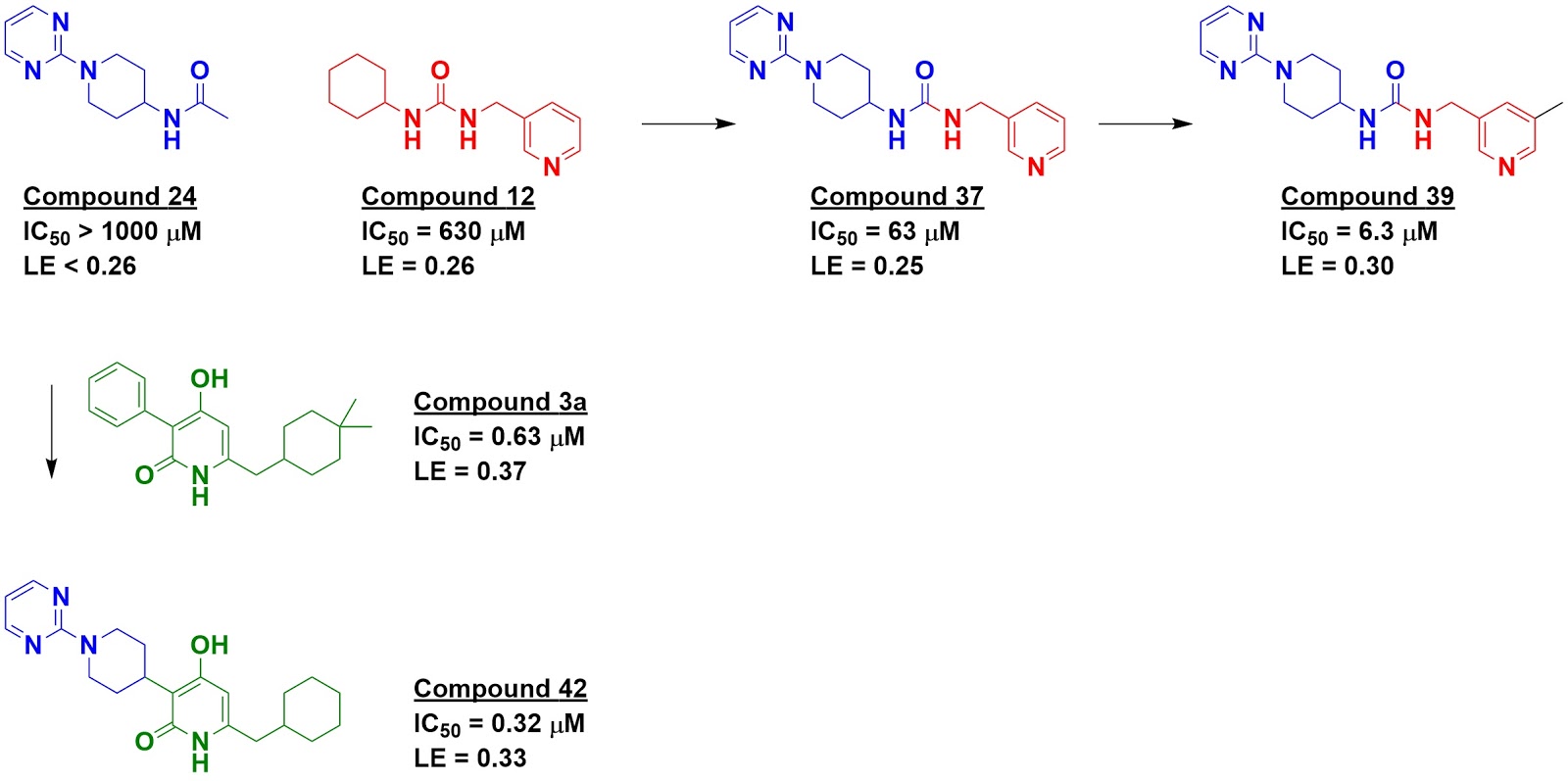
to InhA, including two of the FGC fragments. This information was used to merge
two fragments, compound 24 (an FGC fragment) and compound 12 (a commercial
fragment), yielding a mid-micromolar inhibitor. Adding a “magic methyl” gave a
satisfactory ten-fold boost in potency. Fragment 24 was also merged with a
previously reported molecule, compound 3a, to produce compound 42.
These results suggest that more heavily functionalized fragments don’t
necessarily have a lower hit rate, albeit for a small library and a single
target. And as we noted last year, molecular complexity is difficult to
define; it is not immediately obvious that FGC fragment 24 is actually more complex than commercial fragment 12. The old cliché
still holds: more data are needed.


4 comments:
Hi Dan, while Mike Hann’s molecular complexity model provides an excellent conceptual framework in which to think about molecular recognition and screening library design, its practical value for selection of compounds is limited. An alternative approach is to restrict the extent of substitution and this view of molecular complexity is closer in philosophy to the ‘needle’ concept originating from Roche. While one can restrict extent of substitution directly using SMARTS notation, algorithms such as FLUSH ‘cluster sampling’ tend to pick the most structurally prototypical compound from a group of near neighbors. This is discussed here.
I see the main danger with using fragments that are too small/simple is that you don’t get any hits. Can be useful to assay analogs before starting structural work since it may be possible to get some initial SAR and any additional affinity may help with the structural work. It’s not a big deal that the binding mode for a molecular recognition element evolves with structural elaboration.
I would generally worry about 4-hydroxy-2-pyridones (redox cycling) and the lack of affinity of 42 in the SPR assay would ring a few alarm bells for me.
Hi Pete,
These are good points, and I (somewhat unusually) completely agree.
Thanks too for pointing out the lack of activity for 42 in the SPR screen - that is concerning. What does reassure me is the fact that the closely related molecule compound 3a (NITD-916) has been characterized crystallographically bound to the enzyme (pdb code 4R9S). Also, resistance to NITD-916
develops through mutations in the InhA active site, consisting with direct binding rather than redox cycling.
Anyone know of any studies using DNA-encoded fragments?
good fragment hit rates have also been observed for other complex libraries too. see for instance this paper from researchers at leeds/diamond: 10.1002/chem.201704169
Post a Comment