Three years ago we highlighted a
paper from AstraZeneca arguing for close cooperation of biophysics with high-throughput
screening (HTS) to effectively find genuine hits. A lovely case study just
published in J. Med. Chem. shows just
how beneficial this can be.
Paul Bamborough, Chun-wa Chung,
and colleagues at GlaxoSmithKline and Cellzome were interested in the
bromodomain ATAD2, which is implicated in cancer. (Chun-wa presented some of
this story at the FragNet meeting last year.) Among epigenetic readers,
bromodomains are usually quite ligandable, but ATAD2 is an exception, and when
this work began there were no known ligands.
Recognizing that they might face
challenges, the researchers started by carefully optimizing their protein
construct to be stable and robust to assay conditions. This included screening
1408 diverse compounds, none of which were expected to bind. Disturbingly, a
TR-FRET screen at 10 µM yielded a 4.1% hit rate, suggesting many false positives.
Indeed, when an apparently 30 nM hit from this screen was tested by
two-dimensional 15N-1H HSQC NMR, it showed no binding.
The researchers thus made further refinements to the protein construct to improve
stability and reduce the hit rate against this “robustness set.”
This exercise illustrates an
important point: make sure your protein
is the highest quality possible!
Having done this, the researchers
screened 1.7 million compounds and obtained a relatively modest 0.6% hit rate.
Of these 9441 molecules, 428 showed dose response curves and were tested using
SPR and HSQC NMR. In the case of SPR, the researchers also tested a mutant form
of the enzyme that was not expected to bind to the acetyl-lysine mimics being
sought. Most of the hits did not reproduce in either the SPR or the NMR assays,
and at the end of the process just 16 closely related molecules confirmed – a
true hit rate of just 0.001%!
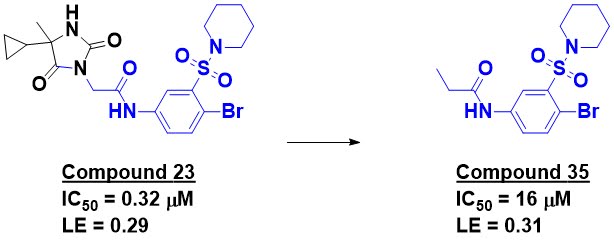
Compound 23 is the most potent
molecule disclosed, but the researchers mention a manuscript in preparation
that describes further optimization. The compound shows promising selectivity
against other bromodomains; it certainly doesn’t look like a classic
bromodomain binder. X-ray crystallography revealed that the binding mode is in
fact different from other bromodomain ligands. Trimming down compound 23
produced compound 35, which shows reasonable activity and ligand efficiency.
This paper nicely demonstrates
the power of biophysics to discern a still small signal in a sea of noise. As
the researchers note, PAINS filters and computational approaches would not have
worked due to the sheer diversity of the misbehaving compounds. (That said, if
the library had been infested with PAINS, the false positive rate would have
been even higher!)
The paper is also a good argument
for FBLD. Compound 35 is probably too large to really qualify as a fragment,
but perhaps related molecules could have led to this series. And GSK also
discovered a different series of potent ATAD2 inhibitors from fragments, which
Teddy wrote about.




