Today’s guest post
is by Glyn Williams (University of Cambridge). Fragment aficionados will
recognize Glyn as the former VP of Biophysics at Astex, but before that he
worked at Roche. His experiences there in the 1990s have lessons for today. -Dan Erlanson
In two recent Practical
Fragments posts (here
and here), Dan
Erlanson noted efforts which will allow the scientific community to contribute
to drug design efforts against the SARS-CoV-2 main protease (Mpro). Leading
the charge at the moment is the COVID
Moonshot consortium who have already received design proposals for covalent
inhibitors, based on the structures of fragments bound to Mpro that
have been generated by researchers at the Diamond Light Source. At the same
time, more information about Mpro, including its substrate
preferences, is being published. Soon there will be an urgent need to define
a selection procedure which will allow valuable drug candidates to be
progressed.
A similar
situation was faced in 1985 when HIV protease was being considered as a drug
target for AIDS. An excellent description of a pragmatic, and ultimately
successful, procedure was published in 1993 by Noel
Roberts and Sally Redshaw of Roche in The Search for Antiviral Drugs:Case Histories from Concept to Clinic.
When the project began there was no definitive proof that this aspartyl protease was essential for viral replication in human cells and that it could not be substituted by a cellular protease. However, its in vitro ability to cleave a Phe-Pro or Tyr-Pro peptide bond (amongst others) marked it out as unusual, and that was sufficient encouragement for Roche to initiate a discovery programme. Inhibitor design then took advantage of this feature to build in selectivity over human aspartyl proteases, ultimately giving a high therapeutic index while also improving inhibitor absorption after oral administration.
Critical issues,
such as the decision to target the HIV-1 viral strain, access to suitable protease
constructs and clear criteria for project progression, were defined early on. Novel
protease and anti-viral assays were then developed in parallel with
transition-state mimetic leads. From the start, it was recognised that the low
aqueous solubility of the optimal peptide substrates could imply that
peptidomimetic inhibitors were also likely to have poor physico-chemical
properties. At the time there was no structural information on the enzyme or
its complexes, so there was little opportunity to avoid these shortcomings.
As with COVID-19, the worldwide health implications of HIV were obvious and scientific interactions between different research groups were driven by a spirit of cooperation. Public laboratories contributed clinical data and provided access to assays for viral activity. In 2020 the ability to share data has improved beyond recognition but the ability to interpret and act on it is still subject to political and commercial pressures. At Roche, a series of hydroxyethylene inhibitors was not pursued due to its prior inclusion in multiple patents for renin inhibitors. In addition, sensitivity to criticism from AIDS activist groups during the project discouraged Roche from developing follow-up candidates later.
Many current predictions and public expectations about COVID-19 now depend on the availability of vaccines in 2021. After more than three decades of research, no preventative vaccine is yet available for HIV. However, the ability to treat a viral infection, even with a drug that contains and controls the infection rather than eliminates it, should not be undervalued. In 1993 the Roche HIV protease clinical candidate, Ro 31-8959, was in Phase 2 evaluation. Roberts and Redshaw pointed out then that lowering a patient’s viral load would reduce the risk of further infections amongst health-care workers and social contacts, while the persistence of immature and non-infectious viral material in cells could stimulate the patient’s own immune system to eliminate the virus.
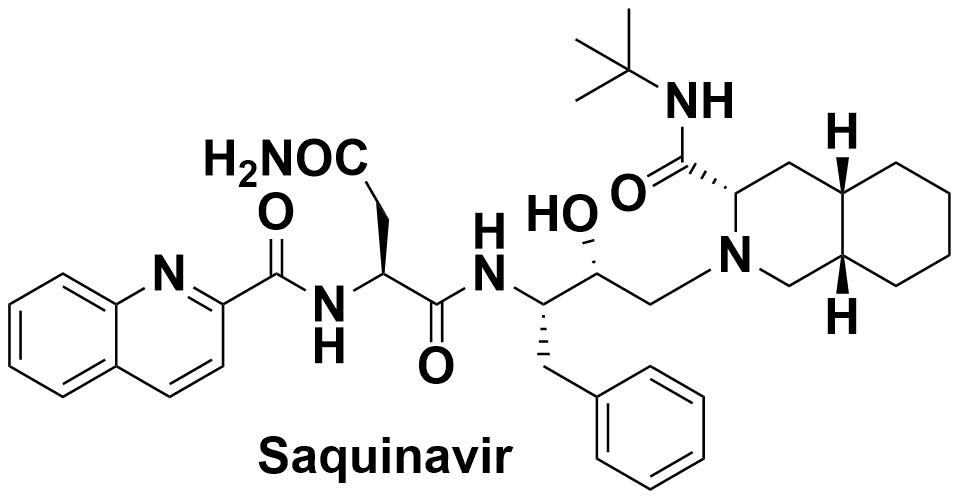 Roberts and
Redshaw concluded their 1993 analysis with the statement that "although there is
still much work to be done, we remain very hopeful that Ro 31-8959 will make a
positive contribution to the therapy of AIDS". Two years later Ro-31-8959, as
Saquinavir, was approved by the FDA and, with Ritonavir, a second protease
inhibitor from Abbott Labs, led to a 64% reduction in deaths from AIDS in the
US over the next 2 years. Let us now hope for the same degree of success from new
COVID-19 treatments.
Roberts and
Redshaw concluded their 1993 analysis with the statement that "although there is
still much work to be done, we remain very hopeful that Ro 31-8959 will make a
positive contribution to the therapy of AIDS". Two years later Ro-31-8959, as
Saquinavir, was approved by the FDA and, with Ritonavir, a second protease
inhibitor from Abbott Labs, led to a 64% reduction in deaths from AIDS in the
US over the next 2 years. Let us now hope for the same degree of success from new
COVID-19 treatments.

7 comments:
Hi Glyn,
I would agree that it’s always good idea in drug discovery (and other scientific endeavours) to see what lessons can be learned from previous studies. For SARS-CoV-2 main protease, I’d be looking at drug discovery efforts against cysteine proteases (e.g. cathepsin K) ahead of the earlier HIV work. Typically, inhibitors of cysteine proteases such as the cathepsins feature a warhead that forms a covalent bond with thiol of the catalytic cysteine and a number of warheads (and scaffolds) have been published in the literature. There are also likely to be large amounts of unpublished inhibition data in industrial databases was well as physical samples in industrial compound collection. Pharma companies have been saying that they want to help and putting some of this data into the public domain would be a good start.
Reversibility is a consideration when designing ligands to bind covalently to a target and I would generally favour ligands that bind reversibly. While irreversible inhibition may be still be a viable way forward, it is important to be fully aware of what you’ll be getting yourself into by going down this route. In particular, assessment of selectivity and PK/PD modelling are both more complex than for reversible inhibitors. I have prepared some notes on reversibility in the context of design of cysteine protease inhibitors.
Hi Pete,
You raise a good point about cysteine proteases, but the unfortunate fact is that to the best of my knowledge no cysteine protease inhibitors have made it all the way through the clinic. Given all the attention on covalent drug discovery in the wake of ibrutinib's success, this is somewhat surprising. Any thoughts as to why, and whether the SARS-CoV-2 main protease could buck the trend?
Hi Pete and Dan,
For me, the central message is that the saquinavir project identified and exploited the unusual features of HIV-1 protease - in that case, its substrate specificity. I would strongly recommend that the current inhibitor design studies for CoV-2 Mpro did the same. The catalytic cysteine of Mpro offers covalent (reversible & irreversible) options for inhibition, but, as Pete points out, practical issues including selectivity, PK/PD, dosage and stability will be major issues. Non-covalent, reversible inhibitors should not be overlooked at this early stage.
I have also not seen any discussion of how any drugs would be used. If the aim is to only treat seriously ill subjects, in a clinical setting and in countries with good access to healthcare, an IV drug will be sufficient. However, Covid-19 is a worldwide infection and so a better strategy may be to limit new infections at source. For this, a selection of oral inhibitors will be required.
Resolving these choices involves socio-political decisions - again, the history of HIV protease offers salient lessons. Scientists should be aware of the wider landscape and understand the long-term implications of their current strategies.
Hi Dan & Glyn,
I believe the cysteine protease inhibitor that went the furthest was the cathepsin K inhibitor odanacatib which choked in phase 3 because of (what I understand to be) some idiosyncratic cardiovascular toxicity. I recall concerns about target-related tox for both cathepsin K and cathepsin L when I worked at AZ on these targets for osteoarthritis. Given the clinical success of HIV and hepatitis C protease inhibitors, I would consider SARS-CoV-2 main protease to be an attractive target (although I’d definitely want to know whether virologists consider this logic to be sound). I would also be interested to know if the enzyme is glycosylated in vivo (this may be an issue for cruzipain since the glycosylation sites are absent in the recombinant form of the enzyme, cruzain, commonly used in inhibition studies). My view is that cysteine proteases are relatively easy to hit if one exploits the catalytic cysteine as a molecular recognition element. For a SARS-CoV-2 main protease project, I would be looking to quickly generate one or two potent probe compounds (they need to be permeable and soluble) in order to investigate their effects in cell-based assays. Significant activity in the cell-based assays would increase confidence in SARS-CoV-2 main protease as a therapeutic target.
I completely agree that one should try to exploit any unusual/unique features of a target in design. There is a wealth of structural information (and associated SAR) for cysteine proteases and this would be a good place to start looking for unusual/unique features of SARS-CoV-2 main protease. This article shows how one can exploit structural differences to modulate selectivity and I’m sure there are plenty of other similar articles in the literature. I favour reversible, covalent inhibition simply because I believe that covalent inhibition represents the quickest way to discover something with the required efficacy and that development of reversible inhibitors is likely to be less complex.
I see IV infusion as the best way to treat acute, life-threatening conditions. The drug clearly needs to be soluble (more so than for an orally-dosed agent) and permeable but you can probably get away with higher clearance than would be the case for an orally-dosed agent. IV infusion is also better for managing tox because you don’t get the variation in plasma levels that you would for an orally-dosed agent. That said, if you have an effective IV agent then you’re in a strong position to identify an oral agent. The key question is what the COVID-19 landscape will look like when a drug is ready for clinical trials. If there is a successful vaccine then it may prove difficult to recruit patients.
Hi Pete,
Thanks for the insightful and interesting comments.
The serine protease of HCV turned out to have a much less-druggable active site than HIV protease and this was ultimately reflected in the macrocyclic nature of the inhibitors and the 20-year timescale required to discover them. Some earlier lead compounds included reversible covalent warheads but the marketed drugs, like Simeprevir, exploit another unusual feature of the protease: inhibition by its product. This was done by including an acyl sulphonamide which acts as a carboxylate isostere. It is also worth noting that the HCV protease inhibitors are dosed orally, so the presence of the macrocycle or acyl sulphonamide did not preclude this route of administration.
My supposition is that other elements of the covalent HCV protease inhibitors were optimised first, before the warhead was added. Dan will have more insight into alternative (and fragment-based) approaches and I fully agree that prior experience will be invaluable, whichever approach is explored. Whatever elements are required to generate efficacy, there should always be advantages in ensuring that the reactivity of any warhead is low and can be geometrically constrained. This should help to achieve the selectivity and in-vivo stability that will be required for a marketed drug.
In the case of HIV, confidence in the value of the protease as a target came from in vitro and in-vivo mutational studies of HIV gag-pol protein processing. This was obtained after the project had started, but before any active compounds were available. The essential nature of the main protease (Mpro) of earlier coronaviruses has already been established but I believe it still needs to be demonstrated for SARS-CoV-2.
In a fast moving field, I am relying on noticeboards to alert me to new developments, so I was grateful for your recent LinkedIn post which highlighted the review by Ghosh et al in ChemMedChem (https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/cmdc.202000223). I have also enjoyed Derek Lowe’s commentaries in his blog ‘In the Pipeline’ (https://blogs.sciencemag.org/pipeline/). As well as his recent post on Covid-19 genetic variations that are being revealed in sequencing data from patients, Derek has highlighted some of the politics involved - including the prominence given to recent trials of Hydroxychloroquine - which can threaten to derail the current scientific progress.
My personal hope is that a currently-licenced, IV or oral, medicine or combination will reduce mortality and infectivity of the virus so that it can be safely treated in hospitals. This would be followed in 12-18 months by a vaccine against SARS-CoV-2, which was then made available globally. That would give more time to find small molecule inhibitors which could (potentially) be redirected to emergent strains or related viruses as required. For that to happen though, there will need to be some financial incentives as well as an absence of political penalties.
What about approved kinase inhibitors that include a cysteine warhead like an acrylamide? Do they give any more confidence in going after Mpro?
Hi Joe,
It is a good question. The approved acrylamide drugs target active-site cysteine residues in one or more of 3 Tyrosine kinases (BTK, EGFR and Her2). They are all approved for cancer therapies, where their significant side effects can be tolerated or managed. On the more positive side, they are all oral drugs with at least some free drug detectable in plasma. This has encouraged work on other cysteine-reactive compounds for cancer targets, like the Ras mutant (G12C), and one of these (AMG510) has recently entered clinical trials.
There has also been a lot of recent work to evaluate less-electrophilic, often reversible-covalent, warheads that would be a better match for targets like Mpro, containing more reactive cysteines. Of particular interest to this audience, a paper on the design and screening of covalent fragment libraries was published by Gyorgy Keseru’s group a few days ago (https://doi.org/10.1016/j.drudis.2020.03.016).
While the molecular understanding of MPro and other SARS-CoV-2 targets is proceeding steadily, the clinical understanding is progressing rapidly. It now seems likely that the highest viral load and infectivity occurs at the very start of the infection and that by the time patients present at hospitals, there is substantially less virus to treat. The damage is done and the subsequent course depends on other factors.
Investigational drugs like Remdesivir may need to be dosed at a very early stage to be effective. If this is confirmed, the identification of inhalable compounds may also be significant. One such compound, which uses a reversible, alpha-ketoamide warhead has been described recently: L. Zhang et al., Science 10.1126/science.abb3405 (2020).
Post a Comment