[**Programming Note** Sorry about two posts in one day, but I thought this was too cool to wait.]
The 3D-arity of fragments is a common topic of discussion in this field. ICYMI, Chris recently did an analysis of PPI interactions and the compounds that target them. However, even more recently, Chris just put up his analysis of the 3DFrag consortium's fragment collection. The 3DFrag collection does not look any more 3D than commercially available collections. Justin Bower from the Beatson points out that this is because their fragment collection is largely due to culling from commercial collections.
Now, no one will argue that nPMI is the best metric for assessing 3D-arity. But it is the best we have so far. So, Chris has tried to improve on the visualization of nPMI for very large libraries. Chris has divided the PMI plot into regions that are disc, rod-like, or spheres (he details how he classifies them at his page). The upshot of this is that he can then generate very simple plots like this:
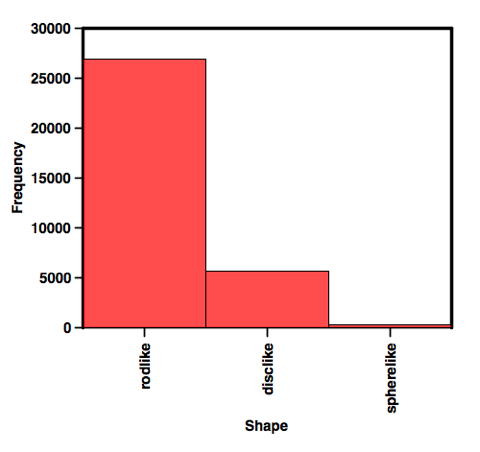
I think this is a great leap forward. Obviously, because of the nature of the chemistry performed over the past umpteen years, this would be totally expected. As Peter Kenny has pointed out previously, rods have volume, but I think that is not what the 3D-eers are aiming at. What's really nice is that 3DFrag has chemists to make fragments. In May, they reported that they had added 221 synthesized fragments. I would like to see how these 221 fragment differ from the commercially available ones. The proof is always in the pudding after all.


8 comments:
I'm not convinced this is the great leap forward that you assert. When you make continuous data categorical, you actually reduce your data-analytic options. Also you need to demonstrate that any conclusions that you arrive at are not dependent on the binning scheme. Visualization of data is often (ususually?)a useful precursor to analysis but you have to be very careful about making decisions based purely on visualization. The other thing to remember is that shape is conformation-dependent.
For the smaller fragments pmi seems a reasonable way to categorise shape, as you get more rotatable bonds it becomes less useful. However from a pragmatic point I've not seen any dramatically better alternatives.
This work tries to help the situation when dealing with the comparison of large collections of fragments. When you plot the points on a graph the overlap of points descends into a blur making comparisons impossible. Binning into rodlike, disclike or spherelike allows you to make the comparison, but I would not use it for anything else.
Pete, I agree it doesn't solve the problem. However, I think it is a substantial step forward. I think of it as more of a guide to making good choices rather than a rule on how to make those decisions.
I've never bought the "it's a guideline not a rule" argument. Too much arm-waving.
I think that it would be possible to sample a space defined using PMI and sampling space is the basis of library design. This sampling could be done using multiple conformations for molecules. If two conformations of one molecule are selected in the library design then so be it. The shape indexing ( dx.doi.org/10.1021/j100011a016 ) that we used in the some of the AZ fragment library design used multiple conformations to represent molecules.
When talking about shape it is important to remember that two molecules (e.g. benzoic acid and benzamidine in their ionized forms) have very similar shapes and yet look very different from the view point of a protein target.
Wrong ref in previous comment (although still worth a look). Here is the correct one: dx.doi.org/10.1021/ci049651v
Chris Richardson of BioFocus gave an interesting talk at the last Cambridge Cheminformatics meeting on Shape-based profiling of fragment collections using "Cube fingerprints". I don't think it has been published yet though.I think they also use multiple conformations of molecules.
Fingerprint-based methods are typically used to define similarity measures which means that a molecular structure is seen in terms of the other molecular structures. This can be described as a distance geometric view of chemical space. One challenge for molecular modeling is to describe molecular recognition in an N-dimensional space. One can do a bit with size and lipophilicity but these don't really capture molecular recognition in a manner that it useful for molecular design.
Although people tend to focus on '3D-ness' the real issue is molecular shape diversity. My own view is that PMI addresses this issue more effectively that either aromatic ring count (which Dan correctly pointed out in this blog a couple of years back is a measure of molecular size) or Fsp3 (which was used to illustrate correlation inflation).
As several approaches to describe 3D-ness have been already mentioned here (Fsp3, PMI), what do the experts think of the recently publsihed "plane of best fit (PBF)"? Reference:
http://dx.doi.org/10.1021/ci300293f
Post a Comment