One of the key advantages of fragment-based drug discovery is that, since there are fewer fragments than lead-sized or drug-sized molecules, it is possible to sample chemical space far more efficiently with fragments than with larger molecules. At least, that’s the theory, but does is it hold true in the real world?
To put it another way, do fragments sample all of the space in which drugs are found? And what kinds of fragments are best for this sampling? In the most recent issue of J. Med. Chem., Stephen Roughley and Rod Hubbard of Vernalis address such questions.
The system they investigate, heat shock protein 90 (Hsp90), is an ideal model system: it is both a popular anti-cancer target as well as structurally tractable, and is thus arguably the most heavily explored single target in terms of fragment-based lead discovery. At least 8 antagonists have entered the clinic, of which at least 2 have come from fragments (see the posts on AT13387, NVP-BEP800/VER-82576, and posts on Evotec compounds discovered by fragment growing or linking.)
Vernalis has had a long-running fragment-based program targeting Hsp90, which has resulted in numerous fragments whose binding modes have been determined by X-ray crystallography. Roughley and Hubbard analyzed these fragments and compared them to published inhibitors. Just 5 distinct fragments can be mapped onto all of the clinical compounds: a handful of fragments effectively samples relevant chemical space. As the authors put it:
For Hsp90 at least, the fragments do cover an appropriate chemical space; what is then important is the imagination of the chemist in evolving the fragments into potent inhibitors.
The second point – about the imagination of the chemist – is critical. Mapping fragments onto elaborated molecules is easier to do retrospectively than prospectively; a cynic could argue that methane is a fragment of just about any drug out there. However, Roughly and Hubbard also point out that, particularly in cases such as this where there are many co-crystal structures, fragments can help identify bioisosteres, including cryptic ones that would not be obvious purely from studying functional SAR.
The paper also addresses the issue of optimal library design, in particular the dilemma of size. Although all five representative fragments were found in an initial set of just 719 fragments, subtle changes can dramatically change the binding mode, an issue we’ve touched on previously. It may not be practical to have multiple similar fragments present in a primary screening library, but testing close analogs after identifying initial fragment hits is likely to be worthwhile.
Finally, one of the concerns about fragment-based approaches is that, if everyone is buying the same set of fragments from the same suppliers and screening them against the same targets, they will end up in the same place – and stumbling over each others’ intellectual property. Reassuringly, this turns out not to be the case:
Even though the various companies discovered rather similar compounds from a fragment screen, exploiting similar binding motifs, there were no exact matches. [Also], the subsequent evolution of the fragments sometimes took very different paths and produced mostly very different chemical leads and candidates.
If this holds true for such heavily mined targets as Hsp90 (and kinases, as discussed previously) it should be even more true for newer classes of targets.
There is a wealth of information in this paper, and it is worth perusing, especially if you find yourself longing for some science over the long holiday weekend.
30 June 2011
26 June 2011
Ligand efficiency and related metrics (Poll)
The discussion following the recent post on LLEAT got me thinking that metrics could be a good topic for a poll (see right side of page).
Click below to see definitions or references, and vote on the right side of the page. Note that you can choose multiple answers.
Antibacterial efficiency
Binding efficiency index (BEI)
Fit quality (FQ)
Fsp3
Ligand efficiency (LE)
Ligand-efficiency-dependent lipophilicity (LELP)
Ligand lipophilic efficiency (LLE)
LLEAT
%LE
Percentage efficiency index (PEI)
Surface-binding efficiency index (SEI)
Other
None
Also, if you use any additional metrics or want to be more specific about how or why you use (or don’t use) the above metrics, please comment.
Click below to see definitions or references, and vote on the right side of the page. Note that you can choose multiple answers.
Antibacterial efficiency
Binding efficiency index (BEI)
Fit quality (FQ)
Fsp3
Ligand efficiency (LE)
Ligand-efficiency-dependent lipophilicity (LELP)
Ligand lipophilic efficiency (LLE)
LLEAT
%LE
Percentage efficiency index (PEI)
Surface-binding efficiency index (SEI)
Other
None
Also, if you use any additional metrics or want to be more specific about how or why you use (or don’t use) the above metrics, please comment.
20 June 2011
Ligand Lipophilicity Efficiency AT Astex Therapeutics
Our last post discussed the growing plague of molecular obesity, and how numerous metrics have been designed to control it. In a paper published online in J. Comput. Aided Mol. Des. Paul Mortenson and Chris Murray of Astex describe a new one: LLEAT.
Although ligand efficiency (LE) is probably the most widely used and intuitive metric, it does not take into account lipophilicity. Other indices do, notably ligand lipophilicity efficiency (LLE) and ligand-efficiency-dependent lipophilicity (LELP), but these both have drawbacks for evaluating fragments. LLE (defined as pIC50 – log P) is not normalized for size; for a fragment to have an (attractive) LLE ≥ 5 it would need an exceptionally low log P or an exceptionally high affinity. LELP, defined as log P / ligand efficiency, is also potentially misleading since a compound could have an acceptable LELP value even with a low ligand efficiency if the log P is also very low.
To address these problems, Mortenson and Murray have tried to strip out the non-specific binding a lipophilic molecule experiences when going from water to a binding site in a protein. They define this modified free energy of binding as:
ΔG* = ΔG - ΔGlipo
≈ RT ln (IC50) + RT ln (P)
≈ ln (10) * RT (log P - pIC50)
In order to put values coming out of this metric on the same scale as those from ligand efficiency, they add a constant, such that:
LLEAT = 0.11 – ΔG* / (number of heavy atoms)
Thus, just as in ligand efficiency, the goal is for molecules to have LLEAT ≥ 0.3 kcal/mol per heavy atom.
The index has some interesting implications. For example, the two fragments below have the same number of heavy atoms, and thus if they had the same activity they would have the same ligand efficiency; on this measure alone, neither would be preferred as a starting point for further work. However, because of their very different lipophilicities, fragment 2 would need to be 45 times more potent than fragment 1 in order to have the same LLEAT of at least 0.3.
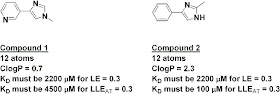 A similar analysis can be done during optimization. For example, adding either a phenyl or a piperazinyl substituent should produce a 20-fold boost in potency in order to maintain ligand efficiency at 0.3, since both have 6 atoms. However, in order to maintain LLEAT at 0.3, the phenyl would need to produce a 460-fold boost in potency while the piperazinyl would need to improve potency only 3-fold. This is consistent with what other folks have reported qualitatively, but it’s nice to have a simple quantitative measure.
A similar analysis can be done during optimization. For example, adding either a phenyl or a piperazinyl substituent should produce a 20-fold boost in potency in order to maintain ligand efficiency at 0.3, since both have 6 atoms. However, in order to maintain LLEAT at 0.3, the phenyl would need to produce a 460-fold boost in potency while the piperazinyl would need to improve potency only 3-fold. This is consistent with what other folks have reported qualitatively, but it’s nice to have a simple quantitative measure.
Although some people may groan at yet another index, and no metric is perfect, I like the fact that this one is intuitive and has the same range of “acceptable” values as ligand efficiency. What do you think – is it useful?
Although ligand efficiency (LE) is probably the most widely used and intuitive metric, it does not take into account lipophilicity. Other indices do, notably ligand lipophilicity efficiency (LLE) and ligand-efficiency-dependent lipophilicity (LELP), but these both have drawbacks for evaluating fragments. LLE (defined as pIC50 – log P) is not normalized for size; for a fragment to have an (attractive) LLE ≥ 5 it would need an exceptionally low log P or an exceptionally high affinity. LELP, defined as log P / ligand efficiency, is also potentially misleading since a compound could have an acceptable LELP value even with a low ligand efficiency if the log P is also very low.
To address these problems, Mortenson and Murray have tried to strip out the non-specific binding a lipophilic molecule experiences when going from water to a binding site in a protein. They define this modified free energy of binding as:
ΔG* = ΔG - ΔGlipo
≈ RT ln (IC50) + RT ln (P)
≈ ln (10) * RT (log P - pIC50)
In order to put values coming out of this metric on the same scale as those from ligand efficiency, they add a constant, such that:
LLEAT = 0.11 – ΔG* / (number of heavy atoms)
Thus, just as in ligand efficiency, the goal is for molecules to have LLEAT ≥ 0.3 kcal/mol per heavy atom.
The index has some interesting implications. For example, the two fragments below have the same number of heavy atoms, and thus if they had the same activity they would have the same ligand efficiency; on this measure alone, neither would be preferred as a starting point for further work. However, because of their very different lipophilicities, fragment 2 would need to be 45 times more potent than fragment 1 in order to have the same LLEAT of at least 0.3.
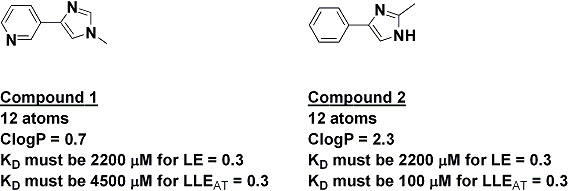
Although some people may groan at yet another index, and no metric is perfect, I like the fact that this one is intuitive and has the same range of “acceptable” values as ligand efficiency. What do you think – is it useful?
12 June 2011
Beware molecular obesity
Obesity in humans is a growing problem, and not just aesthetically: the condition may be responsible for millions of premature deaths. In a recent article in Med. Chem. Commun., Mike Hann of GlaxoSmithKline notes that “molecular obesity” is also leading to the untimely demise of far too many drug development programs.
Hann, who is especially known for his work on molecular complexity, defines molecular obesity as the “tendency to build potency into molecules by the inappropriate use of lipohilicity.” This is the result of an unhealthy “addiction” to potency. Hann suggests that since potency is easy to measure it is pursued preferentially to other factors, particularly early in a program. This is all too often achieved by adding mass, much of it lipophilic. The problem is that all this grease decreases solubility and increases the risks of off-target binding and toxic side effects.
Tools such as the Rule of 5 have been developed in part to avoid this problem, and a number of other indices have been introduced more recently. For example, lipophilic ligand efficiency (LLE) is defined as pIC50 – LogP; molecules with an LLE > 5 are likely to be more developable. Other guidelines that Hann mentions and that have been covered here include LELP, number of aromatic rings, and fraction of sp3 hybridized carbon atoms. But this is not to say that metrics will save the day:
Hann also argues that potency itself is over-rated: many teams seek single digit nanomolar binders even though approved drugs have average potencies of 20 nM to 200 nM.
The paper is a fun read (and is free after registration too). Also, Hann one-ups Donald Rumsfield’s (in)famous “known knowns, known unknowns, and unknown unknowns” by pointing out that much of this information falls into the category of “unknown knowns”:
Hann, who is especially known for his work on molecular complexity, defines molecular obesity as the “tendency to build potency into molecules by the inappropriate use of lipohilicity.” This is the result of an unhealthy “addiction” to potency. Hann suggests that since potency is easy to measure it is pursued preferentially to other factors, particularly early in a program. This is all too often achieved by adding mass, much of it lipophilic. The problem is that all this grease decreases solubility and increases the risks of off-target binding and toxic side effects.
Tools such as the Rule of 5 have been developed in part to avoid this problem, and a number of other indices have been introduced more recently. For example, lipophilic ligand efficiency (LLE) is defined as pIC50 – LogP; molecules with an LLE > 5 are likely to be more developable. Other guidelines that Hann mentions and that have been covered here include LELP, number of aromatic rings, and fraction of sp3 hybridized carbon atoms. But this is not to say that metrics will save the day:
The problem with the proliferation of so many “rules” is the trend to slavishly apply them without really understanding their required context for use and subsequent limitations.Starting a program with the smallest possible lead should in theory lead to smaller drugs, and this is one of the key justifications for fragment-based approaches, though even here it is important that the molecules do not become obese during optimization. One of the themes at the 6th annual FBDD conference was to do a bit of optimization around the fragment itself before growing or linking. On a related note, Rod Hubbard warned in his opening presentation at the conference to “beware the super-sized fragment.” Not only are larger fragments likely to be less complementary to the target, the number of possibilities increases (and thus the coverage of chemical space drops) by roughly ten-fold with each atom added to a fragment.
Hann also argues that potency itself is over-rated: many teams seek single digit nanomolar binders even though approved drugs have average potencies of 20 nM to 200 nM.
The paper is a fun read (and is free after registration too). Also, Hann one-ups Donald Rumsfield’s (in)famous “known knowns, known unknowns, and unknown unknowns” by pointing out that much of this information falls into the category of “unknown knowns”:
Those things that are known but have become unknown, either because we have never learnt them, or forgotten about them, or more dangerously chosen to ignoreThis review is an excellent corrective to the first two problems, and a clear warning about the third.