Virtual screening is continuing
to make impressive strides. The latest example, in Nature, comes from William
Wetsel (Duke), John Irwin (UCSF), Georgios Skiniotis (Stanford), Brian Shoichet
(UCSF), Bryan Roth (UNC Chapel Hill), Jonathan Ellman (Yale), and a large group
of collaborators. The paper has received considerable attention (for example In
the Pipeline), but in my opinion the connection to FBLD has been understated.
The researchers were interested
in finding new agonists for the 5-HT2A receptor (5-HT2AR).
This GPCR is the target for LSD and psilocybin, both of which have been shown
to reduce depression and anxiety. Is it possible to find molecules with similar
therapeutic activity but without the accompanying psychedelic properties?
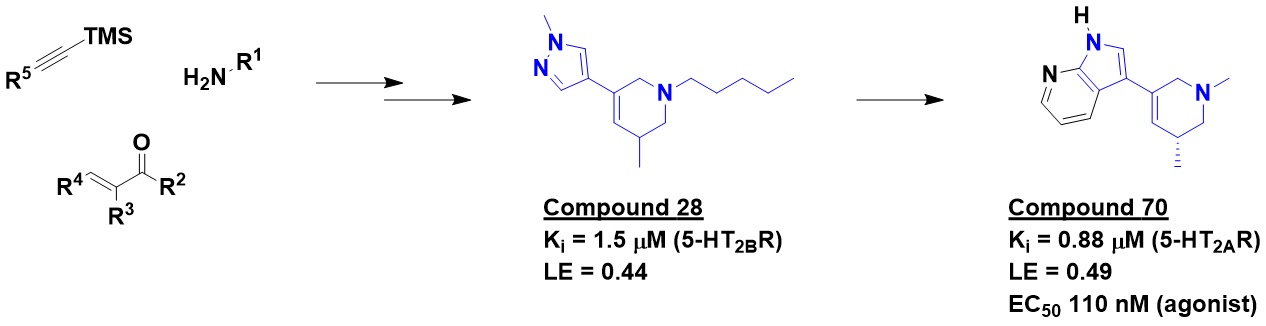
LSD contains a tetrahydropyridine
(THP) moiety, which is relatively rare in screening libraries. The researchers developed
convergent routes to THPs in which they could independently and efficiently
vary multiple substituents. Using this chemistry, they constructed a virtual library
of 4.3 billion compounds, all with molecular weights ≤ 400 Da and cLogP ≤ 3.5.
At the time the research began,
there were no structures of 5-HT2AR, so the researchers built a
homology model based on the closely related 5-HT2BR, which differs
by only four amino acid residues in the orthosteric pocket where LSD binds. This
model was then screened against a subset of the THP library, those ≤ 350 Da.
Despite screening some 7.45 trillion complexes (sampling an average of 92 conformations
and 23,000 orientations per molecule), the process took only nine hours on a
1000-core CPU cluster. The result was 300,000 hits in nearly 15,000 families. To
ensure novelty, only compounds quite different from known ligands were further considered,
and 17 “richly functionalized” THPs were synthesized and tested in radioligand
assays. Four were active, including racemic compound 28. Searching the 4.3
billion compound library for analogs ultimately led to compound 70 and a related,
slightly more potent molecule lacking the methyl substituent on the amine. A
cryo-EM structure subsequently validated the predicted binding mode.
The paper spends considerable
time characterizing these two compounds. Both are agonists and somewhat
selective for 5-HT2AR over 5-HT2BR and 5-HT2CR.
They are highly selective over 318 other GPCRs and 45 off-targets. GPCRs can
signal through arrestin and/or G-protein, and while LSD works (mainly) through
the arrestin pathway, the new molecules work (mainly) through the G-protein
route. Importantly, the compounds showed anti-depressive and anti-anxiety
effects in mouse models. Although you can’t ask mice if they are tripping, the
molecules did not cause “head-twitch responses” and other behavioral effects seen
with LSD, suggesting that they may not have hallucinogenic properties.
This is a lovely piece of work, and
a few observations relevant to FBLD stand out. First, the best molecules are
actually rule-of-three compliant, despite the fact that larger molecules were
included in the virtual screen. Indeed, the top two molecules are actually smaller
than the initial hits. This suggests that choosing more richly functionalized
molecules may not have been the most efficient approach. We’ve written
previously about V-SYNTHES, which entails stepwise selection and growing of
fragments; it would be interesting to retroactively test whether this type of
approach would have more quickly gotten to compound 70.
Finally, this approach can easily
be extended to other scaffolds for which syntheses are readily available. Six
years ago we wrote about the synthetic accessibility of dihydroisoquinolines, and
last year Practical Fragments published our fifth “fragment library
roundup.” The marriage of clever chemistry with virtual screening seems to have
a bright future.
No comments:
Post a Comment