We’ve previously discussed the proliferation of metrics used to evaluate fragments. Ligand efficiency is by far the most popular, and what it and most other measurements have in common is that they represent binding affinity (or inhibition, or some other surrogate). Binding affinity is associated with thermodynamics – how well a molecule binds to a target – but this measure says nothing about how rapidly a molecule associates and dissociates from the target (kinetics). In the November issue of Drug Discovery Today Geoffrey Holdgate and Adrian Gill at AstraZeneca propose a new metric, kinetic efficiency (KE), to address this issue:
KE = τ / (# of heavy atoms) = t1/2 / (0.693 * (# of heavy atoms))
where τ is the residence time or relaxation constant and is, in the simplest case, 1/koff
koff is the dissociation rate constant
and t1/2 is the half-life for dissociation
Why are the kinetics of dissociation important? Holdgate and Gill list a series of drugs for hypertension and note that compounds that remain bound to the receptor longer avoid rapid clearance and thus have superior clinical activity. On the other hand, drugs for schizophrenia that bind the D2 dopamine receptor can cause side effects if they remain bound too long. Thus, optimal kinetic efficiency is case-dependent .
Though kinetics of ligand binding can be assessed with techniques like SPR, this parameter is often ignored. However, as Holdgate and Gill point out, slow-binders are likely to be lead-sized or drug-sized molecules. Indeed, none of the roughly two-dozen examples they present would satisfy the rule of 3.
This raises an interesting question: how often do fragments dissociate slowly? Slowly-dissociating fragments are often flagged as pathological in SPR studies. Intuitively it seems that smaller molecules would have faster kinetics; a small fragment is likely to be able to dart in and out of a protein-binding site more rapidly than a larger molecule that requires some movement on the part of the protein to accommodate its binding. Still, there must be some cases of fragments with slow dissociation rate constants. If you know of any please mention them in the comments section.
28 November 2011
18 November 2011
And once more into the breach...
When the market is more than 20 Billion dollars, you will find everyone working there. And so, with this recent publication, we have another entrant into the BACE inhibitor from Fragments competition, discussed previously here. This is the fifth by my count, the first being from Astra Zeneca.
In this paper, Eli Lilly describes their efforts using fragments to generate "the first orally available non-peptidic BACE1 inhibitor that produces profound Abeta-lowering effects in animals." They screened ~8000 compounds at 4.76mM that generated a number of low-affinity, but highly "LEAN" fragments (discussed below). Of most interest were the amino-benzothiazine (1) and amino-thiadiazine (2) compounds.

The authors note that co-crystallilzation was a key advance for their understanding of this system. The co-crystal showed two copies of (1) with high active site occupancy and in the "open-flap" conformation. One copy engaged the catalytic dyad and the other spanned the S1-S3 cavity. This data let them recognize that the planarity of the molecules were not optimal for fragment growth, so they "de-planarized" them, leading to (3). Only one enantiomer of (3) was recognized by BACE. The co-crystal of this compound showed binding identical to the original fragment, one copy engaging the catalytic dyad and one in the S1-S3 region. Addition of the S3 moiety pyrimidine led to (4). Fluorination of the central ring reduced in vivo clearance and and realized a significant increase in potency, while maintaining atom efficiency (5).
 The crystal structure of (5) shows that this molecule retains an optimal H-bonding network, efficiently traverses S1, and projects the pyrimidine into S3.
The crystal structure of (5) shows that this molecule retains an optimal H-bonding network, efficiently traverses S1, and projects the pyrimidine into S3.
Compound (5) was tested in animal models and pre-clinically in healthy human volunteers given orally. It showed significant reduction in Abeta levels in brain and CSF. Retinal pathology became a concern in longer term animal studies and the compound was not taken any further.
This paper shows the power of Fragments in discovering novel scaffolds for important targets. It is also important to note that the modified fragment hit retained the same binding as the original fragment hit.
The other contribution that the Lilly group brings out in this paper is the concept of LEAN (Ligand Efficiency by Atom Number): -log (IC50)/Number of heavy atoms. This is one of many ways people have developed to gauge the efficiency of their ligand hits, I think this is the simplest to use. As can be seen from the Lilly data, a LEAN of >=0.30 is an efficient molecule. For those of us who don't do logs in our head well, this lends it itself to a simple cheat sheet:
 I can send a copy of this spreadsheet to anyone who wants.
I can send a copy of this spreadsheet to anyone who wants.
In this paper, Eli Lilly describes their efforts using fragments to generate "the first orally available non-peptidic BACE1 inhibitor that produces profound Abeta-lowering effects in animals." They screened ~8000 compounds at 4.76mM that generated a number of low-affinity, but highly "LEAN" fragments (discussed below). Of most interest were the amino-benzothiazine (1) and amino-thiadiazine (2) compounds.

The authors note that co-crystallilzation was a key advance for their understanding of this system. The co-crystal showed two copies of (1) with high active site occupancy and in the "open-flap" conformation. One copy engaged the catalytic dyad and the other spanned the S1-S3 cavity. This data let them recognize that the planarity of the molecules were not optimal for fragment growth, so they "de-planarized" them, leading to (3). Only one enantiomer of (3) was recognized by BACE. The co-crystal of this compound showed binding identical to the original fragment, one copy engaging the catalytic dyad and one in the S1-S3 region. Addition of the S3 moiety pyrimidine led to (4). Fluorination of the central ring reduced in vivo clearance and and realized a significant increase in potency, while maintaining atom efficiency (5).
 The crystal structure of (5) shows that this molecule retains an optimal H-bonding network, efficiently traverses S1, and projects the pyrimidine into S3.
The crystal structure of (5) shows that this molecule retains an optimal H-bonding network, efficiently traverses S1, and projects the pyrimidine into S3.Compound (5) was tested in animal models and pre-clinically in healthy human volunteers given orally. It showed significant reduction in Abeta levels in brain and CSF. Retinal pathology became a concern in longer term animal studies and the compound was not taken any further.
This paper shows the power of Fragments in discovering novel scaffolds for important targets. It is also important to note that the modified fragment hit retained the same binding as the original fragment hit.
The other contribution that the Lilly group brings out in this paper is the concept of LEAN (Ligand Efficiency by Atom Number): -log (IC50)/Number of heavy atoms. This is one of many ways people have developed to gauge the efficiency of their ligand hits, I think this is the simplest to use. As can be seen from the Lilly data, a LEAN of >=0.30 is an efficient molecule. For those of us who don't do logs in our head well, this lends it itself to a simple cheat sheet:
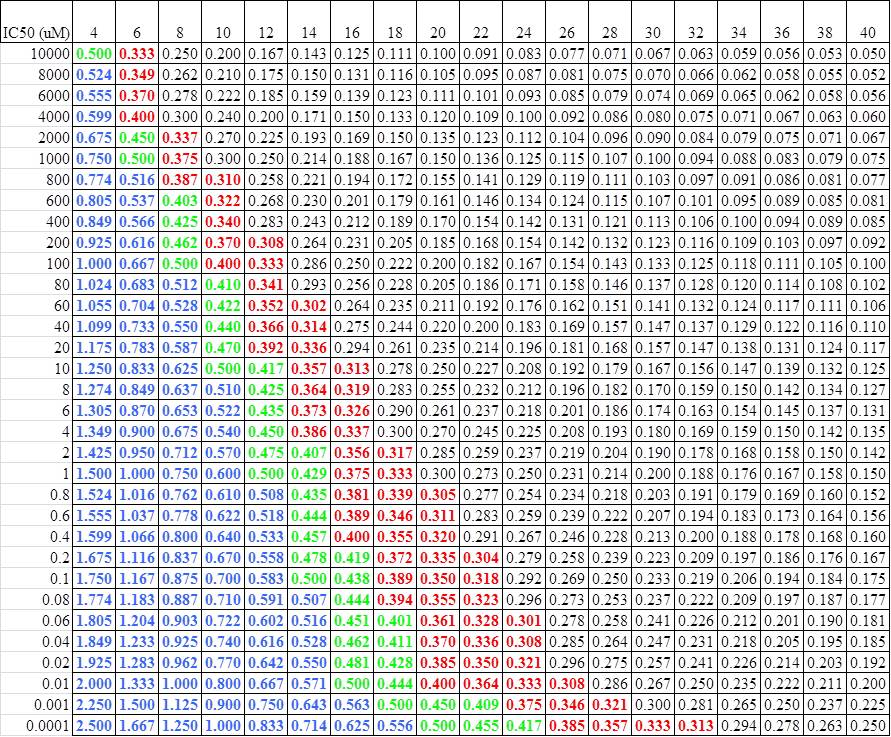 I can send a copy of this spreadsheet to anyone who wants.
I can send a copy of this spreadsheet to anyone who wants.
17 November 2011
What do fragment hits look like?
Our last post highlighted a study showing that most of the best fragment hits loosely followed the Rule of 3, even though the library from which they were selected was not strictly Rule of 3 compliant. As it happens, Chris Swain at Cambridge MedChem Consulting has been tabulating fragment hits reported in the literature and has assembled a database of more than 280. Previously he has assessed the physicochemical properties of commercially available libraries; now, he’s analyzed the fragments that have actually been reported as hits and has published the results here.
For the most part the fragments conform to the Rule of 3. Size-wise most of them are truly fragments, with the majority having molecular weights less than 250 Da. Not surprisingly, they also tend to be fairly aromatic. Interestingly, roughly one third of the fragments are charged at physiological pH, with a pretty even split between acids and bases.
Of course, despite the overall Rule of 3 compliance, there are outliers in all the parameters, especially hydrogen-bond acceptors. So perhaps, to paraphrase Darren Begley channeling Bill Murray, the Rule of 3 should be renamed the Guideline of 3.
For the most part the fragments conform to the Rule of 3. Size-wise most of them are truly fragments, with the majority having molecular weights less than 250 Da. Not surprisingly, they also tend to be fairly aromatic. Interestingly, roughly one third of the fragments are charged at physiological pH, with a pretty even split between acids and bases.
Of course, despite the overall Rule of 3 compliance, there are outliers in all the parameters, especially hydrogen-bond acceptors. So perhaps, to paraphrase Darren Begley channeling Bill Murray, the Rule of 3 should be renamed the Guideline of 3.
10 November 2011
Pushing the Rule of 3
The Rule of 3 (Ro3) is commonly used to design fragment libraries. First published as a brief 450-word (shorter than this post!) “update” in the discussion forum of Drug Discovery Today in 2003 by researchers at Astex, it has become the fragment equivalent of Chris Lipinski’s famous Rule of 5. Like that rule, it has its critics, notably our friends at FBDD and Molecular Design. A key point of contention is whether the Ro3 is too restrictive. A new paper in J. Med. Chem. from Gerhard Klebe’s group at Philipps University Marburg addresses this question.
The definition of the Rule of 3 provided by Astex is as follows:
The study indicated that such hits seem to obey, on average, a ‘Rule of Three’, in which molecular weight is <300, the number of hydrogen bond donors is ≤3, the number of hydrogen bond acceptors is ≤3 and ClogP is ≤3. In addition, the results suggested NROT (≤3) and PSA (≤60) might also be useful criteria for fragment selection.
One of the criticisms leveled at the Ro3 is that it is vague in terms of what constitutes a hydrogen bond acceptor. For example, does the nitrogen in an amide count? What about the nitrogen in an indolizine? Presumably for simplicity Lipinski assumed that any nitrogen or oxygen atom would count as a hydrogen bond acceptor. At the risk of engaging in exegesis, I propose that only oxygen or nitrogen atoms most medicinal chemists would consider as acceptors should be counted as acceptors, and that the limits on the number of rotatable bonds (NROT) and polar surface area (PSA) are optional.
In the recent paper, which is also discussed at FBDD and Molecular Design, Klebe and colleagues assembled a library of 364 fragments in which the average properties of the fragments were within Ro3 guidelines (with the exception of “Lipinski acceptors,” which would include the nitrogen of a tertiary amide), but there were some outliers. They then performed a fluorescence-based competition screen against the model protein endothiapepsin, resulting in 55 fragments that inhibited at least 40% at 0.5 or 1 mM concentration. These fragments were taken into crystallography trials, resulting in 11 structures. The paper presents lots of nice analysis of how these fragments bind to the protein. It also notes that:
Only 4 of the 11 fragments are consistent with the rule of 3. Restriction to this rule would have limited the fragment hits to a strongly reduced variety of chemotypes.
This may be an overstatement. Looking at the fragment hits more closely, all of them have molecular weights less than 300, and only one has ClogP > 3. Personally, given the problems of molecular obesity and the dangers of lipophilicity, I’d say that these aspects of the Ro3 are the most important, and find it notable that the hits were so compliant given that the library did contain larger, more lipophlic members.
All 11 of the crystallographically characterized fragments also have 3 or fewer hydrogen bond donors and TPSA < 60 Å2. Only two of the fragments have more than 3 rotatable bonds, but where the majority of the fragments fail to pass Ro3 is in the number of “Lipinski acceptors,” where 6 of the 11 have > 3. However, if you count hydrogen bond acceptors more judiciously (ie, compound 291 would have 3 acceptors rather than 4, since the aniline nitrogen would not be counted), only 1 of the 11 fragments has more than 3 acceptors.
Like most rules, the Rule of 3 should never be treated as a strait-jacket. That said, given the number of possible small fragment-sized molecules, and the necessarily limited size of any fragment collection, there seems to be plenty of room within the Rule of 3 for attractive chemical diversity.
The definition of the Rule of 3 provided by Astex is as follows:
The study indicated that such hits seem to obey, on average, a ‘Rule of Three’, in which molecular weight is <300, the number of hydrogen bond donors is ≤3, the number of hydrogen bond acceptors is ≤3 and ClogP is ≤3. In addition, the results suggested NROT (≤3) and PSA (≤60) might also be useful criteria for fragment selection.
One of the criticisms leveled at the Ro3 is that it is vague in terms of what constitutes a hydrogen bond acceptor. For example, does the nitrogen in an amide count? What about the nitrogen in an indolizine? Presumably for simplicity Lipinski assumed that any nitrogen or oxygen atom would count as a hydrogen bond acceptor. At the risk of engaging in exegesis, I propose that only oxygen or nitrogen atoms most medicinal chemists would consider as acceptors should be counted as acceptors, and that the limits on the number of rotatable bonds (NROT) and polar surface area (PSA) are optional.
In the recent paper, which is also discussed at FBDD and Molecular Design, Klebe and colleagues assembled a library of 364 fragments in which the average properties of the fragments were within Ro3 guidelines (with the exception of “Lipinski acceptors,” which would include the nitrogen of a tertiary amide), but there were some outliers. They then performed a fluorescence-based competition screen against the model protein endothiapepsin, resulting in 55 fragments that inhibited at least 40% at 0.5 or 1 mM concentration. These fragments were taken into crystallography trials, resulting in 11 structures. The paper presents lots of nice analysis of how these fragments bind to the protein. It also notes that:
Only 4 of the 11 fragments are consistent with the rule of 3. Restriction to this rule would have limited the fragment hits to a strongly reduced variety of chemotypes.
This may be an overstatement. Looking at the fragment hits more closely, all of them have molecular weights less than 300, and only one has ClogP > 3. Personally, given the problems of molecular obesity and the dangers of lipophilicity, I’d say that these aspects of the Ro3 are the most important, and find it notable that the hits were so compliant given that the library did contain larger, more lipophlic members.
All 11 of the crystallographically characterized fragments also have 3 or fewer hydrogen bond donors and TPSA < 60 Å2. Only two of the fragments have more than 3 rotatable bonds, but where the majority of the fragments fail to pass Ro3 is in the number of “Lipinski acceptors,” where 6 of the 11 have > 3. However, if you count hydrogen bond acceptors more judiciously (ie, compound 291 would have 3 acceptors rather than 4, since the aniline nitrogen would not be counted), only 1 of the 11 fragments has more than 3 acceptors.
Like most rules, the Rule of 3 should never be treated as a strait-jacket. That said, given the number of possible small fragment-sized molecules, and the necessarily limited size of any fragment collection, there seems to be plenty of room within the Rule of 3 for attractive chemical diversity.