Most readers of this blog are familiar with the concept of ligand efficiency:
LE = (free energy of ligand binding) / (number of heavy atoms)
The metric is easy to calculate, intuitive, and useful for evaluating fragments. Weirdly, as ligands get larger, the ligand efficiencies tend to decrease. This is a consequence of the fact that the maximal affinities of ligands also start to plateau as their sizes increase, a fact noted more than a decade ago by Kuntz and colleagues. The reason for this has never been satisfactorily explained, but the effect is so pronounced that some researchers have proposed alternatives to LE that take it into account, for example %LE and fit quality. In a paper published online in ACS Med. Chem. Lett., Charles Reynolds and M. Katherine Holloway have delved into the origins of declining LE with increasing size in more detail.
The researchers analyzed 102 ligand-protein complexes representing 14 target classes for which thermodynamic data had been collected using isothermal titration calorimetry (ITC). They calculated ligand efficiency as well as “enthalpy efficiency” and “entropy efficiency”, where enthalpy or entropy takes the place of free energy in the numerator. In the case of enthalpy efficiency, the trend was the same as for ligand efficiency: as molecules got larger, the enthalpy efficiencies tended to decrease. For entropy efficiencies, however, there was essentially no trend. In other words, the leveling out of binding energy is due to enthalpic effects more than to entropic effects. The authors suggest that:
The size effect on enthalpy is a result of increasing ligand complexity and the need to satisfy multiple geometric constraints simultaneously.
The findings thus provide further support for trying to identify fragments whose binding is largely enthalpically driven, but, as usual in the real world, things are not quite so easy. One of the frustrating things about medicinal chemistry is the phenomenon of enthalpy-entropy compensation: if you try to improve the enthalpy of binding, say by adding a hydrogen-bond acceptor, you may end up paying an entropic penalty such that overall binding increases only modestly, if at all. Indeed, the data in the paper show a very strong correlation between enthalpy and entropy: enthalpic binders tend to have negative (unfavorable) entropy, and the most entropic binders actually have positive (highly unfavorable) enthalpies of binding.
Worse, despite the striking correlation between enthalpy and entropy, there is almost no correlation between free energy of binding and enthalpy or entropy. The researchers suggest that these perverse trends (or lack thereof) explain why computational approaches to drug discovery are not more successful: most modeling relies on one or a few low-energy conformations, in effect ignoring entropy. However, there is a correlation between overall binding energy and enthalpy for certain targets, such as HIV protease and aldose reductase; perhaps such targets are more amenable to modeling.
In summary, the message seems to be that you should try to find enthalpic binders if you can, though the binding energetics may change unpredictably during optimization. For modelers, the research suggests that ignoring entropy is unwise. For medicinal chemists, I’m not sure how much of this is actionable, but sometimes it’s nice to know the origins behind the perversity of the universe.
19 May 2011
Fragments vs Pim-1
The three members of the Pim family of serine/threonine kinases help cancer cells proliferate and survive, and are thus interesting as potential anti-cancer targets. Structurally the kinases are also intriguing because they have a proline residue in the “hinge” region of the purine binding site, differentiating them from the other 500+ kinases in the human genome. Two recent papers describe different fragment-screening approaches against Pim-1: one case rediscovers fragments of a previously reported compound and the other identifies a new series of potent inhibitors unlike other reported kinase inhibitors.
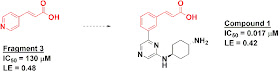
In the first paper, published in Acta Cryst. Section D, researchers from Bayer Schering Pharma AG performed a crystallographic screen of just 36 fragments against Pim-1. Despite the small library size, a dozen of the fragments produced interpretable electron density, although only 4 of these showed activity better than 10 mM. Fragment 3, with an IC50 of 130 micromolar in an enzymatic assay, was the most potent. Interestingly, a close analog of this cinnamic acid fragment was also part of a Pim-2 inhibitor previously identified through HTS by a group from Boehringer Ingelheim. The Bayer folks synthesized this molecule (compound 1), confirmed that it is also potent against Pim-1, and found that, crystallographically, it overlays beautifully with the fragment.
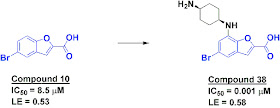
 The second paper, published in Bioorg. Med. Chem. Lett., describes how researchers at Genzyme used a fragment-based approach to develop potent Pim-1 inhibitors. The researchers used SPR to screen a library of about 1800 fragments at 75 micromolar concentration and then used a biochemical assay to characterize the active molecules. Benzofuran-2-carboxylic acids such as compound 10 (below) were particularly interesting: not only did they have high ligand efficiencies, they don’t look anything like typical kinase inhibitors. X-ray crystallography revealed that the bromine atom is binding in a hydrophobic pocket, while the acid is making hydrogen bond contacts with the catalytic lysine and other residues. A related fragment with comparable potency had a methoxy substituent in the 7-position, and by transforming this into a positively charged moiety the researchers were able to improve the potency to low nanomolar with a dramatic increase in ligand efficiency.
The second paper, published in Bioorg. Med. Chem. Lett., describes how researchers at Genzyme used a fragment-based approach to develop potent Pim-1 inhibitors. The researchers used SPR to screen a library of about 1800 fragments at 75 micromolar concentration and then used a biochemical assay to characterize the active molecules. Benzofuran-2-carboxylic acids such as compound 10 (below) were particularly interesting: not only did they have high ligand efficiencies, they don’t look anything like typical kinase inhibitors. X-ray crystallography revealed that the bromine atom is binding in a hydrophobic pocket, while the acid is making hydrogen bond contacts with the catalytic lysine and other residues. A related fragment with comparable potency had a methoxy substituent in the 7-position, and by transforming this into a positively charged moiety the researchers were able to improve the potency to low nanomolar with a dramatic increase in ligand efficiency.
 What’s also interesting is that if you overlay the two fragments from the two papers, they superimpose almost exactly on top of one another, with the carboxylic acid moieties making the same interactions – clearly a hot spot.
What’s also interesting is that if you overlay the two fragments from the two papers, they superimpose almost exactly on top of one another, with the carboxylic acid moieties making the same interactions – clearly a hot spot.
 On a side note, as most readers are probably aware, sanofi-aventis has recently acquired Genzyme. Practical Fragments wishes all the folks in Massachusetts well during the integration.
On a side note, as most readers are probably aware, sanofi-aventis has recently acquired Genzyme. Practical Fragments wishes all the folks in Massachusetts well during the integration.
In the first paper, published in Acta Cryst. Section D, researchers from Bayer Schering Pharma AG performed a crystallographic screen of just 36 fragments against Pim-1. Despite the small library size, a dozen of the fragments produced interpretable electron density, although only 4 of these showed activity better than 10 mM. Fragment 3, with an IC50 of 130 micromolar in an enzymatic assay, was the most potent. Interestingly, a close analog of this cinnamic acid fragment was also part of a Pim-2 inhibitor previously identified through HTS by a group from Boehringer Ingelheim. The Bayer folks synthesized this molecule (compound 1), confirmed that it is also potent against Pim-1, and found that, crystallographically, it overlays beautifully with the fragment.



16 May 2011
Native MS: turning up the voltage
At the FBDD meeting in San Diego last month there was some discussion of the connection between protein-ligand binding in the gas phase (ie, in native mass-spectrometry experiments) vs in solution; the discussion continued in the comment section here. Valerie Vivat, head of the mass spectrometry, molecular & cell biology department at NovAliX, has now provided a thorough response.
Valerie makes some interesting points here, in particular the statement that "the gas phase stability is not an indication of the complex binding affinity." Still, I do like the fact that native MS could be used as a quick way to sort enthalpic from entropic binders. What do you think?
When analyzing non-covalent complexes by mass spectrometry under non-denaturing conditions, one must keep in mind indeed how interactions stabilizing the complex in solution will be affected by the ion transfer in the gas phase. Basically, interaction based on hydrophobic effect is lost while the electrostatic-based interactions (Van der Waals, H-bonds, ionic interaction) are preserved (and are even strengthened due to the absence of solvent shielding). Regarding water molecules now, during the ESI process, biomolecules are transferred in the gas phase as partially hydrated ions and complete ion desolvation is subsequently achieved through low energy collisions with the residual gas molecules in the mass spectrometer interface. This complete desolvation is required for accurate mass measurements. Consequently, in most cases, molecular mass measurement of the complexes is accurate enough to rule out the possibility that water molecules remain after ions are transferred from the atmospheric pressure to the deep vacuum of the TOF analyzer, even in the case of crystallographic water molecules.
Regarding gas phase stability : Reports in the literature mention that collision-induced dissociation experiments and the determination of Vc50s can be used to monitor gain or loss in polar interaction. Indeed, we have now accumulated a variety of in house examples showing that increase in the gas phase stability of a complex is correlated with a gain in polar interactions as shown by X-ray or ITC. Monitoring the complex gas phase stability is thus an attractive feature to quickly evaluate, for example, gain or loss in polar interaction of analog series targeting the same protein binding site.
Importantly, the gas phase stability is not an indication of the complex binding affinity. Evaluation of complex affinity (after complexes are formed in solution) is done under instrumental conditions showing no dissociation of the non-covalent complexes. For complexes stabilized by electrostatic-based interactions (and thus rather stable in the gas phase), it is indeed much easier to find optimal instrument settings compared to complexes stabilized by hydrophobic effect. However, this does not preclude complexes mainly stabilized in solution by hydrophobic effect to be detected at all. For example, nuclear hormone receptors in complex with fatty acids or phospholipids are readily detected with appropriate settings. In this case, the polar head of the ligands is likely to act as a stable anchor in the gas phase, resulting in non-covalent complexes which remain intact during the ca. 1 millisecond flight of the ion from the ionization source to the detector.
Importantly, the gas phase stability is not an indication of the complex binding affinity. Evaluation of complex affinity (after complexes are formed in solution) is done under instrumental conditions showing no dissociation of the non-covalent complexes. For complexes stabilized by electrostatic-based interactions (and thus rather stable in the gas phase), it is indeed much easier to find optimal instrument settings compared to complexes stabilized by hydrophobic effect. However, this does not preclude complexes mainly stabilized in solution by hydrophobic effect to be detected at all. For example, nuclear hormone receptors in complex with fatty acids or phospholipids are readily detected with appropriate settings. In this case, the polar head of the ligands is likely to act as a stable anchor in the gas phase, resulting in non-covalent complexes which remain intact during the ca. 1 millisecond flight of the ion from the ionization source to the detector.
To sum up, native MS can be run under two different modes:
1. Low energy conditions where no complex dissociation occurs are used to assess the binding affinity of protein / ligand complexes,
2. High energy conditions inducing complex dissociation provide insight into the extent of polar interaction involved between the protein and the ligand. This type of experiment makes sense to compare a series of molecules binding to the same protein binding site.
Valerie makes some interesting points here, in particular the statement that "the gas phase stability is not an indication of the complex binding affinity." Still, I do like the fact that native MS could be used as a quick way to sort enthalpic from entropic binders. What do you think?
11 May 2011
Fragments vs ion channels
Ion channels represent an intriguing class of targets for a variety of indications, but it is difficult to develop robust high throughput assays. The gold standard is to actually measure the electrical conduction of the channels using techniques such as patch clamping, but this is time-consuming and tedious. Unfortunately, faster indirect methods are prone to high rates of false positives. Medium throughput automated patch clamp technologies are now available, and in a recent issue of Bioorg. Med. Chem. Lett. Scott Wolkenberg and colleagues at Merck decided to use one of these for a fragment screen.
The researchers screened 1280 fragments against the sodium ion channel acid-sensing ion channel 3 (ASIC3), a potential target to treat pain. Compounds were screened at 1 mM concentration, and 56 of them inhibited >50%. Interestingly, this 4.4% hit rate is comparable to other fragment screens at Merck on completely different target classes. Dose-response curves were generated for the 56 compounds, yielding 32 hits in 12 separate chemical series with ligand efficiencies > 0.3. Similarity searching led to the testing of 250 analogs, but most of these were less active than the original fragments.
In the absence of structural information, two strategies were used to try to improve the potencies. First, a series of 220 analogs were synthesized in which small (approximately 64 mass units) substituents were systematically added at various positions around the fragments. Unfortunately, most of the compounds were inactive, and those that were active were only marginally better than the starting fragment. The second strategy was to make more dramatic changes using parallel chemistry of up to a few hundred compounds. This was more successful: for example, fragment 3 led to compound 24, with 200-fold improved activity, albeit with a loss in ligand efficiency.
 Unfortunately the best compounds reported still do not break the micromolar barrier, and the pharmacokinetics are not encouraging. The authors conclude that:
Unfortunately the best compounds reported still do not break the micromolar barrier, and the pharmacokinetics are not encouraging. The authors conclude that:
The researchers screened 1280 fragments against the sodium ion channel acid-sensing ion channel 3 (ASIC3), a potential target to treat pain. Compounds were screened at 1 mM concentration, and 56 of them inhibited >50%. Interestingly, this 4.4% hit rate is comparable to other fragment screens at Merck on completely different target classes. Dose-response curves were generated for the 56 compounds, yielding 32 hits in 12 separate chemical series with ligand efficiencies > 0.3. Similarity searching led to the testing of 250 analogs, but most of these were less active than the original fragments.
In the absence of structural information, two strategies were used to try to improve the potencies. First, a series of 220 analogs were synthesized in which small (approximately 64 mass units) substituents were systematically added at various positions around the fragments. Unfortunately, most of the compounds were inactive, and those that were active were only marginally better than the starting fragment. The second strategy was to make more dramatic changes using parallel chemistry of up to a few hundred compounds. This was more successful: for example, fragment 3 led to compound 24, with 200-fold improved activity, albeit with a loss in ligand efficiency.

The present results are consistent with the analysis that advancing fragment hits to high potency (<100 nM) and/or to a preclinical development milestone in the absence of structural information is unlikely.I think this may be overly pessimistic. While optimizing fragments in the absence of structure is certainly challenging, it is possible. Fragment screening ultimately delivered several distinct series of low micromolar compounds against ASIC3. Had these come from an HTS assay, the job of optimizing them further would not be any easier. Moreover, this report represents another example of using fragment approaches to tackle membrane proteins.