One of the things I did at FBLD2012 was count how many fragments were in the libraries people were reporting on. Now, we all know that libraries are dynamic and not static, but I thought the numbers would give an idea of how big libraries are across the industry (and academia). I may have missed some, but these are the ones I caught:
Pfizer: 3000-5000 cpds @30mM in DMSO with 50-100 mg of powder in supply
GSK: 2400 cpds @100mM in DMSO. ~14 HA average for their "regular" library
~1000 cpds for NMR purposes
~1500 19F cpds
Selcia (Not sure if this is their library or the one they ran for a client, somebody with better notes?)
1500 cpds @30mM DMSO, purity >95%
Roche: 6900 cpds
Elan: 5260 cpds
Heptares: 4500 cpds, average HAC 16
Amgen: 1200 19F cpds
ZoBio: 1500 cpds
CSIRO: 480 cpds
Emerald ~2000 cpds
U Cambridge: 1300 cpds
Genentech: 2500 cpds
Ariad 735 Ro3 compliant cpds
Vanderbilt: 11000 cpds
Abbott: ~10000 cpds
Merck: ~10000 cpds
Plexxikon: 20000 scaffolds (explicitly differentiated from fragments in the talk)
28 September 2012
26 September 2012
FBLD 2012
Fragment-based Lead Discovery 2012 concluded today in San Francisco. This is
the fourth in an illustrious series of conferences that started in San Diego in 2008, moved to York
in 2009, and then to Philadelphia
in 2010. These meetings set a high bar in terms of quality, and I think it’s
fair to say that FBLD 2012 has lived up to its predecessors. With 39 talks, 16
vendors, over 50 posters, and close to 250 attendees I won’t attempt a
comprehensive summary, so please weigh in if you were there.
Pete Kenny is always ahead of the curve, and so despite his
absence from the conference he did put together a nice preview. He worried that
the big pharma talks might be “strategy-heavy and results-light,” but happily
this was not the case. In particular, Jane Withka gave a very detailed talk on
Pfizer’s carefully constructed fragment library (see also here). One statistic
that caught my eye is that, after screening this library against 21 proteins,
44% of the 2500+ fragments have hit at least one target – considerably higher
than the 33% that has been seen in several organizations. Whether this is
because of a better library or simply more screens remains unclear.
Fragment validation – or the lack thereof – and fragment
promiscuity were also frequently recurring topics. Peter Kutchukian from
Novartis observed that frequent hitters are not always problematic: fragments
that hit multiple targets were actually more
likely to produce co-crystal structures than more selective fragments.
There were lots of cool approaches for finding fragments;
one particularly impressive advance was presented by SensiQ in a workshop
before the main conference. Their surface plasmon resonance (SPR) instrument
operates as expected; what sets it apart is a cunningly designed injection
method in which the sample compound flows through a long capillary before
entering the flow cell. A concentration gradient forms within the capillary,
and by controlling this gradient the user can run a full dose-response curve
over several orders of magnitude without
having to pre-dilute the sample. Instead of doing a primary screen at a
fixed concentration and then following up on active compounds, dissociation
constants can be determined directly from a primary screen.
Protein flexibility is a subject dear to my heart, but not
typically observed in biophysical fragment-finding techniques besides
crystallography and protein-detected NMR. Josh Salafsky described Biodesy’s
second-harmonic generation technology to specifically find molecules that cause
conformational changes. More surprisingly, Beactica’s Helena Danielson argued
that SPR could be used to observe conformational changes in membrane proteins. Although
I’m no SPR expert, I've only seen the technique being used to detect changes in mass, so it will be fun to see how this develops.
Fluorine NMR looks like it’s finally coming into
its own; Brad Jordan discussed how this has become a standard screening
technique at Amgen, and both Nino Campobasso (GlaxoSmithKline) and Stephan Zech
(Ariad) mentioned that their companies are using it too.
In addition to the method talks there were plenty of
hit-to-lead and success stories, some of which have been covered on Practical
Fragments, and some of which will be as the publications come out.
If you weren’t able to make it this year, FBLD 2014 is
tentatively planned to be held in Basel,
Switzerland.
And if you can’t wait that long for your next fragment fix, there is at least
one more relevant event this year and several already taking shape for 2013 –
details to come shortly.
21 September 2012
Fragments vs DDAH – covalent and noncovalent
Using biochemical assays to find fragments sidesteps the
need for expensive biophysical instruments, but is fraught with difficulties.
In a recent paper in Bioorg. Med. Chem.,
Thomas Linsky and Walter Fast at the University of Texas, Austin, describe how
they used functional screening to discover fragment inhibitors of
dimethylarginine dimethylaminohydrolase (DDAH), an enzyme involved in nitric
oxide production.
The researchers used a 4000-member fragment library from ChemBridge,
which they screened against two different isoforms of DDAH at 0.1 mM in a
biochemical screen. This resulted in 79 hits against the human enzyme and 44
hits against a bacterial (P. aeruginosa)
DDAH, 101 in total. 66 of these were then repurchased along with 41 analogs,
and tested in a completely different biochemical assay against human DDAH-1; 31
showed >20% inhibition at 0.4 mM, including only 22 of the original hits,
suggesting that many of the initial hits were indeed false positives. Further
studies showed that 5 of the 31 compounds interfered with this secondary assay (ie,
they showed “inhibition” even in the absence of enzyme), suggesting that they were
false positives in both the primary and secondary biochemical assays.
DDAH contains an active-site cysteine, and the researchers wanted
to exclude molecules that might be generically reactive, so they incubated the
remaining 26 compounds with the low-molecular weight thiol glutathione and then
retested them; this eliminated another 21 compounds.
Finally, the remaining 5 compounds were examined by
mass-spectrometry, and one of these turned out to be a compound other than what
was listed on the bottle! This left just 4 legitimate fragment hits.
Two of these compounds were 4-halopyridines, which, although
not generally reactive with thiols, could covalently modify the active site
cysteine of DDAH (see here for more details). The other two compounds were
reversible, competitive inhibitors of human DDAH-1. Although low affinity (Ki
values of 0.8 and 1.7 mM), they had respectable ligand efficiencies (0.38 and
0.29 kcal/mol/atom, respectively).
Interestingly, when Linksy and Fast retrospectively analyzed
where the four validated fragments came from, they found that only the two
halopyridines were detected in the primary screen; the two reversible fragment
hits had been purchased for the secondary round of screening as analogs of
primary hits.
This is a richly detailed and well-executed example of
fragment-based screening in academia. It demonstrates once again that
high-concentration biochemical screens can be used to find fragments, but be
prepared to wade through a lot of junk: only about 2% of the original hits
proved to be legitimate (see here for similar results from Vernalis on a different
target). It also illustrates the utility of exploring analogs of initial
fragment hits; in this case, even though most of the primary hits didn’t hold
up, they nonetheless led to new fragments. Of course, this does raise the
question of how spurious primary hits can lead to genuine inhibitors – what do
you think?
14 September 2012
Fragments vs CYPs – on purpose
The cytochrome P450 enzymes, or CYPs, are a huge class of
oxidizing enzymes found across all kingdoms of life. In humans these enzymes metabolize
many drugs, and to avoid drug-drug interactions, drug hunters generally shun or
re-engineer molecules that inhibit CYPs. But microorganisms such as Mycobacterium tuberculosis (Mtb), the
causative agent of tuberculosis, also contain CYPs, and targeting these could
lead to a sorely needed new treatment for this disease. A team led by Chris
Abell at the University
of Cambridge has
published just such a strategy in Angew.
Chem. Int. Ed.


The researchers were interested in CYP121, which is unique
to Mtb and important for its viability. They used a thermal-shift assay to
screen 665 commercial fragments at 5 mM, of which 66 increased the melting
point by at least 0.8 ˚C. 56 of these were further characterized by STD and
WaterLOGSY NMR, and 26 showed interactions with the protein and could also be
competed with a known substrate. Eight of the most soluble of these were then
soaked into crystals of CYP121, leading to four high-resolution structures,
three of which are shown here. Isothermal titration calorimetry was used to
determine dissociation constants.
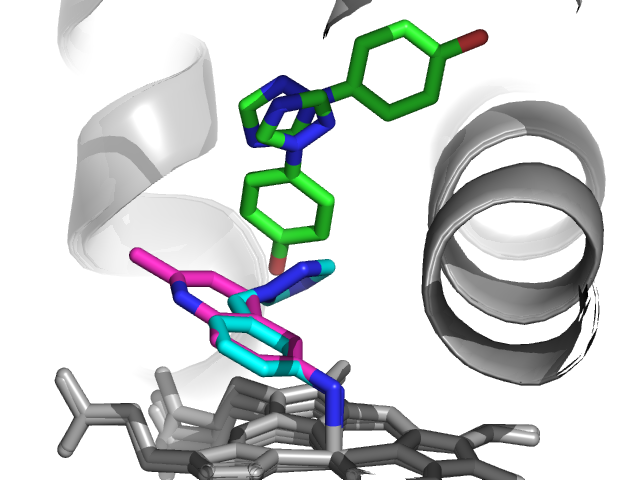
Interestingly, two of these fragments (1 and 2 in blue and
red, respectively) coordinate to the heme iron, while the other two do not.
Fragment 4 (green in figure above) showed two binding modes in the crystal structure,
leading the researchers to make molecules that merge both binding modes.
Although the resulting molecules bind in a similar fashion as compound 4 as
judged crystallographically, they show at best marginal improvements in
affinity and sizable losses in ligand efficiency. Quantum mechanical calculations
suggested that this lack of improvement was due to conformational strain within
the molecules.
Happily, merging compounds 1 and 2 was much more successful,
leading to compound 14, which maintained ligand efficiency and improved
affinity. A crystal structure revealed that, as designed, compound 14 binds in
a very similar manner as the initial fragments. The molecule was also selective
for CYP121 over a different CYP from Mtb as well as several human CYPs.

This is a nice paper not only because it reports a
successful example of fragment merging on a new class of targets, but because
it also describes several approaches that didn’t
work. Fragment merging and fragment linking probably fail more often than they
succeed, and this report really digs into the SAR and addresses why merging can
be so challenging.
Of course, what would be really cool would be to link
compound 14 with compound 4 (ie, link all of the fragments in the top figure),
and the paper ends with the statement that this is currently ongoing. It will
be fun to see the results.
10 September 2012
Fluorine Fetish
As readers of this blog may know, I love 19F NMR. 19F NMR is well known in screening applications, like FAXS (fluorine chemical shift anisotropy and exchange for screening) and FABS (fluorine atoms for biochemical screening). Back in January, I reviewed a very nice paper from Amgen on the subject. Now, Anna Vulpetti and Claudio Dalvit, the father of 19F in industry, have a review out in Drug Discovery Today on Post Screen applications. This review tries to make the case for ubiquitous use of 19F.
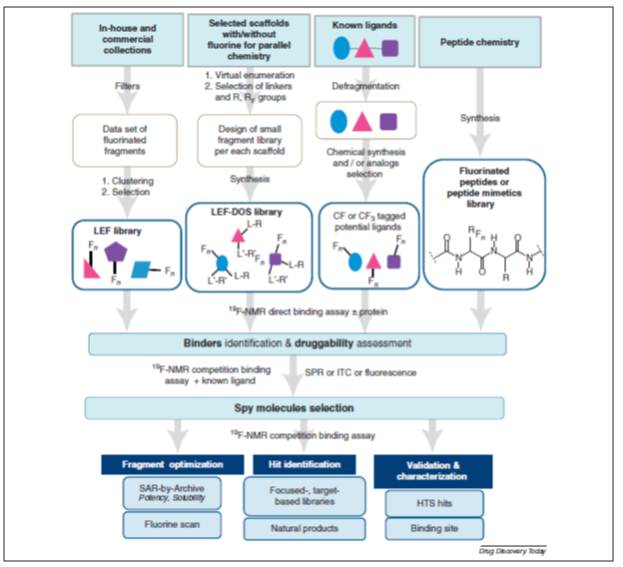 They propose a complete workflow for using 19F NMR (Figure Above). They point out the variety of ways that fluorine can impact the H2L stage. Flourine has many advantages when replacing proton: increased metabolic stability, increased acidity, and decreased basicity. Lipophilicity is increased if the fluorine is near a basic nitrogen or on a aromatic ring. Permeability can be increased. Fluorine can also be a powerful binding moiety by means of H-bond interactions, lipophilic contacts and multipolar and/or antiparallel polar interactions, indirect contact with the protein mediated by water molecules, or by preorganizing the ligand conformation as needed for the protein recognition.
They propose a complete workflow for using 19F NMR (Figure Above). They point out the variety of ways that fluorine can impact the H2L stage. Flourine has many advantages when replacing proton: increased metabolic stability, increased acidity, and decreased basicity. Lipophilicity is increased if the fluorine is near a basic nitrogen or on a aromatic ring. Permeability can be increased. Fluorine can also be a powerful binding moiety by means of H-bond interactions, lipophilic contacts and multipolar and/or antiparallel polar interactions, indirect contact with the protein mediated by water molecules, or by preorganizing the ligand conformation as needed for the protein recognition.However, this leads to a point Dan made almost three years ago:
I wondered why fluorine-labeled fragments are not used more widely; Fluorine has a strong NMR signal and is very sensitive to the local environment, so when a fluorine-containing fragment binds to a protein this can be easily detected. In fact, the dynamic range for this type of assay is so great that fragment binding can be detected at concentrations several orders of magnitude lower than their dissociation binding constants.
This seems like a very powerful approach, but I haven’t seen many other people using it. Are folks concerned about the need for fluorine in every fragment (although many are commercially available) or is there something else I’m missing?
- Not everyone has 19F probe they can dedicate to screening, follow up, etc.
- 19F libraries require a lot of work to put together
- 19F is magic methyl by another name
- See 1.
Let us know your thoughts in the comments.