In addition to dozens of reviews on fragment-based drug discovery, entire books have been published, the first in 2006 and the second in 2008. Now a third has joined the list: Volume 493 of the venerable Methods in Enzymology series, edited by Lawrence Kuo at Johnson & Johnson, is titled Fragment-Based Drug Discovery: Tools, Practical Approaches, and Examples. With 21 chapters and roughly 600 pages, it is a comprehensive addition to the field. Whether you’re just starting out in the field of fragments or already an expert, this is an invaluable resource.
As the subtitle suggests, the book is divided into three sections. The first, Tools, is the shortest, consisting of just 6 chapters. Chapter 1, by Brett Tounge and Michael Parker, describes how they assembled a 900 fragment library at Johnson & Johnson for use in crystallography-based screening. Chapter 2, by Gaetano Montelione and colleagues, discusses the high-throughput protein preparation approach that the Northeast Structural Genomics Consortium has taken, including advice on what to do when problems arise. The next two chapters are devoted to crystallography: Jark Böttcher and colleagues from Proteros Biostructures provide a general overview including practical tips in Chapter 3, while Doug Davies and colleagues from Emerald BioStructures analyze the results of 18 in-house campaigns to try to draw general conclusions of why some targets are more successful than others in Chapter 4. On the subject of difficult targets, Chapter 5 is devoted to GPCRs, specifically the stabilized versions being generated by Miles Congreve and coworkers at Hepatares Therapeutics and analyzed by SPR and TINS NMR in collaboration with researchers at the University of Utah and ZoBio. Finishing out this section, Renee DesJarlais of Johnson & Johnson provides an overview and practical tips for applying computational approaches to FBDD.
The second section, Practical Approaches, begins with a chapter by Lawrence Kuo on how to ensure that hits from fragment screens will be distinct from those coming out of HTS. Chapter 8, by Tony Gianetti at Genentech, is a valuable and comprehensive tutorial on using SPR for fragment screening. The next two chapters are devoted to NMR: Chapter 9, by Christopher Lepre at Vertex, discusses such practicalities as library preparation and NMR screening conditions, while Chapter 10, by Joshua Ziarek and colleagues at the Medical College of Wisconsin provides more detail on specific NMR techniques. The following two chapters discuss a couple less common techniques. Chapter 11, by James Kranz at GlaxoSmithKline and Celine Schalk-Hihi at Johnson & Johnson, gives an excellent description of the protein thermal shift technique for identifying and characterizing fragments, complete with all the mathematics. And Chapter 12, by Lars Neumann and colleagues at Proteros Biostructures, describes the reporter displacement assay and its use to select for fragments with desirable kinetic and thermodynamic profiles. In Chapter 13, John Spurlino of Johnson & Johnson describes crystallography-based fragment screening and advancement without collecting affinity data; this was recently discussed here, but the chapter provides additional details and examples. Finally, the last chapter in this section, by Zenon Konteatis and colleagues at Ansaris, discusses the incorporation of protein flexibility into computational fragment-based screening.
The last section, Examples, opens with a chapter by Masaya Orita and colleagues at Astellas that covers ligand efficiency and related indices and an overview of strategies to advance fragments, with a particular emphasis on “anchor-based drug discovery.” Chapter 16, by James Lanter and colleagues at Johnson & Johnson, emphasizes the importance of engaging medicinal chemists in fragment projects from an early stage. In Chapter 17, Hugh Eaton and Daniel Wyss at Merck describe using NMR in FBDD both in general terms and with respect to their BACE1 program, which has entered clinical trials. Chapter 18, by Till Maurer at Genentech, is also devoted to NMR, focusing on the specific steps involved. Chapter 19, by Marta Abad and colleagues at Johnson & Johnson, returns to crystallography, further discussing the electron-density guided FBDD approach in the context of the target ketohexokinase. In Chapter 20, Rod Hubbard and James Murray discuss a decade of fragment work at Vernalis, integrating many of the techniques discussed in the rest of the volume (as well as a few new ones!) and providing a valuable discussion on why orthogonal biophysical fragment-finding methods are necessary. The book closes with a chapter by Darren Begley and colleagues at Emerald BioStructures and collaborators at the University of Washington on a structural genomics initiative to use crystallography-guided methods to tackle proteins important in infectious diseases.
Although each of these chapters can be downloaded individually, it is well worth getting a copy of the book itself. Like other members of the series, it is beautifully put together. Having all the papers in one place is extremely convenient, and simply paging through the volume is bound to generate new ideas.
29 March 2011
22 March 2011
Fragments vs. PDK1 again: into the adaptive pocket
PDK1 holds a certain appeal for fragment-based approaches. This kinase, an upstream member of the phosphatidylinositol-3 kinase (PI3K) signaling pathway, is an attractive anti-cancer target and has been successfully targeted using FBDD by researchers at Vernalis and GlaxoSmithKline (see here for a very recent publication). A series of allosteric activators was also discovered by researchers at Pfizer. The latest report on this target, an inhibitor which is particularly potent against the unphosphorylated form of PDK1, was just published online in Bioorg. Med. Chem. Lett.; it describes some of the work my colleagues at Sunesis and I did in collaboration with researchers at Biogen Idec.
To find inhibitors against PDK1, we used Tethering with extenders. This approach starts by covalently modifying a protein of interest with an “extender,” which is a fragment designed to bind in a desired site on a protein and to capture other fragments; it contains a protein-reactive functionality as well as a masked thiol. In this case, we used a generic fragment known to interact with the purine-binding pocket of kinases, a diaminopyrimidine (red in upper left of figure). The protein-extender complex was then screened against a library of several thousand disulfide-containing fragments under partially reducing conditions; only fragments with some affinity for the protein will remain covalently bound to the protein and thus be detectable by mass-spectrometry. The pyridinone (blue in figure) was one of the strongest hits.
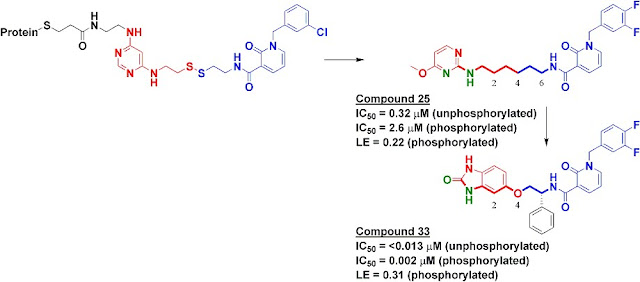
We synthesized several molecules containing purine-pocket binding elements connected to pyridinones by flexible linkers and found compound 25 to be one of the most potent. The SAR revealed the importance of a hydrogen-bond donor-acceptor pair (green atoms in figure) a specific distance from the pyridinone fragment, and further medicinal chemistry led ultimately to compound 33. In addition to being quite potent, compound 33 was remarkably selective for PDK1: in a panel of 241 kinases screened at 10 micromolar concentration of compound 33, only PDK1 and one other kinase were inhibited by 80% or more.
The selectivity of compound 33 was partially explained by the X-ray crystallographic structure, which revealed that the pyridinone fragment discovered through Tethering binds in the so-called adaptive pocket with the activation loop in the “DFG-out” conformation. I believe this is the only series of inhibitors for which this binding mode has been observed for PDK1. Consistent with the structure, these inhibitors are more potent against the unphosphorylated (inactive) form of PDK1 than against the phosphorylated (active) form.
Compound 33 was effective at preventing phosphorylation of the PDK1 substrate Akt both in cells as well as in a xenograft model. Though I may be biased in thinking so, this is a nice application of a fragment-based approach to discover a strikingly specific kinase inhibitor. I hope that people will use compound 33 to further probe the biology of the PI3K pathway. Indeed, at least one completely independent team of researchers seems to already be doing so.
To find inhibitors against PDK1, we used Tethering with extenders. This approach starts by covalently modifying a protein of interest with an “extender,” which is a fragment designed to bind in a desired site on a protein and to capture other fragments; it contains a protein-reactive functionality as well as a masked thiol. In this case, we used a generic fragment known to interact with the purine-binding pocket of kinases, a diaminopyrimidine (red in upper left of figure). The protein-extender complex was then screened against a library of several thousand disulfide-containing fragments under partially reducing conditions; only fragments with some affinity for the protein will remain covalently bound to the protein and thus be detectable by mass-spectrometry. The pyridinone (blue in figure) was one of the strongest hits.
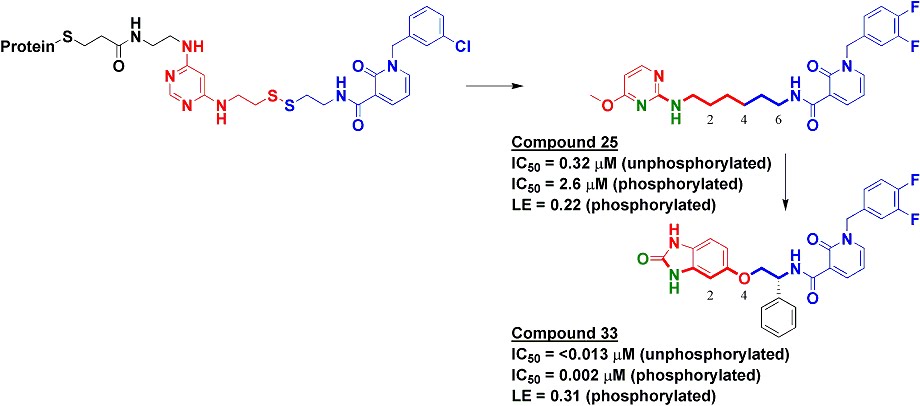
We synthesized several molecules containing purine-pocket binding elements connected to pyridinones by flexible linkers and found compound 25 to be one of the most potent. The SAR revealed the importance of a hydrogen-bond donor-acceptor pair (green atoms in figure) a specific distance from the pyridinone fragment, and further medicinal chemistry led ultimately to compound 33. In addition to being quite potent, compound 33 was remarkably selective for PDK1: in a panel of 241 kinases screened at 10 micromolar concentration of compound 33, only PDK1 and one other kinase were inhibited by 80% or more.
The selectivity of compound 33 was partially explained by the X-ray crystallographic structure, which revealed that the pyridinone fragment discovered through Tethering binds in the so-called adaptive pocket with the activation loop in the “DFG-out” conformation. I believe this is the only series of inhibitors for which this binding mode has been observed for PDK1. Consistent with the structure, these inhibitors are more potent against the unphosphorylated (inactive) form of PDK1 than against the phosphorylated (active) form.
Compound 33 was effective at preventing phosphorylation of the PDK1 substrate Akt both in cells as well as in a xenograft model. Though I may be biased in thinking so, this is a nice application of a fragment-based approach to discover a strikingly specific kinase inhibitor. I hope that people will use compound 33 to further probe the biology of the PI3K pathway. Indeed, at least one completely independent team of researchers seems to already be doing so.
18 March 2011
Weak affinity chromatography (WAC)
There are many ways to find fragments: NMR and X-ray crystallography are old favorites, but SPR is quickly catching on. There are also more specialized approaches, such as ITC, MS, TINS, and biochemical screening. Now another 3-letter abbreviation has joined the list: a paper published online by Sten Ohlson and colleagues at Linnaeus University in Sweden in Analytical Biochemistry describes weak affinity chromatography, or WAC.
The principle is remarkably simple. First, a protein of interest is covalently immobilized onto a chromatography column packed with modified silica gel. This can be done on a standard high-performance liquid chromatograph (HPLC). Then each fragment to be tested is injected in buffer; those that have affinity for the immobilized protein will stay on the column longer than they would if they lacked affinity. The fragments can be detected with either UV spectrometry or mass spectrometry.
To demonstrate the technique, the researchers used two model enzymes, thrombin and trypsin, and a couple dozen fragments ranging in mass from 93 to 307 Da. Most of these fragments contained an amidine, a moiety known to bind to both proteins. Columns without any immobilized protein served as controls. Of course, fragments may associate non-specifically with proteins, so the researchers also treated protein-containing columns with irreversible or potent reversible inhibitors; a fragment that comes out later from a column containing an active protein than from a column containing an inactive protein is presumably binding specifically to the active site.
Remarkably, the technique appears to work: most of the amidine-containing fragments were retarded in columns containing active protein compared to columns containing inhibited protein. Moreover, the relative affinity ranking correlated with the inhibitory activity of fragments in enzymatic assays. Some of the fragments were quite weak, with calculated dissociation constants around 1 mM.
The researchers also demonstrated that they could screen a mixture of 11 fragments, using mass-spectrometry to follow each fragment, and that the change in retention time was comparable to that observed when running each fragment individually. In this case it was important to use low fragment concentrations so as to avoid saturating the protein active sites.
As with any technique, there are bound to be limitations. The immobilized protein needs to be stable for an extended time; in the current case there was some degradation in performance, albeit over the course of months and more than 200 injections. A more serious constraint is the need for a proper reference. Inactivating an enzyme provides an ideal solution, but one that won’t be so easily generalized to all targets.
In some ways WAC could be seen as a low-price cousin of TINS: both methods rely on an immobilized protein, but while TINS uses a custom modified NMR spectrometer, WAC can get by with a much less pricey HPLC (though a mass-spectrometer seems nearly indispensible). It will be fun to see how WAC develops, and in particular whether it can be used to discover novel fragments against more challenging targets.
The principle is remarkably simple. First, a protein of interest is covalently immobilized onto a chromatography column packed with modified silica gel. This can be done on a standard high-performance liquid chromatograph (HPLC). Then each fragment to be tested is injected in buffer; those that have affinity for the immobilized protein will stay on the column longer than they would if they lacked affinity. The fragments can be detected with either UV spectrometry or mass spectrometry.
To demonstrate the technique, the researchers used two model enzymes, thrombin and trypsin, and a couple dozen fragments ranging in mass from 93 to 307 Da. Most of these fragments contained an amidine, a moiety known to bind to both proteins. Columns without any immobilized protein served as controls. Of course, fragments may associate non-specifically with proteins, so the researchers also treated protein-containing columns with irreversible or potent reversible inhibitors; a fragment that comes out later from a column containing an active protein than from a column containing an inactive protein is presumably binding specifically to the active site.
Remarkably, the technique appears to work: most of the amidine-containing fragments were retarded in columns containing active protein compared to columns containing inhibited protein. Moreover, the relative affinity ranking correlated with the inhibitory activity of fragments in enzymatic assays. Some of the fragments were quite weak, with calculated dissociation constants around 1 mM.
The researchers also demonstrated that they could screen a mixture of 11 fragments, using mass-spectrometry to follow each fragment, and that the change in retention time was comparable to that observed when running each fragment individually. In this case it was important to use low fragment concentrations so as to avoid saturating the protein active sites.
As with any technique, there are bound to be limitations. The immobilized protein needs to be stable for an extended time; in the current case there was some degradation in performance, albeit over the course of months and more than 200 injections. A more serious constraint is the need for a proper reference. Inactivating an enzyme provides an ideal solution, but one that won’t be so easily generalized to all targets.
In some ways WAC could be seen as a low-price cousin of TINS: both methods rely on an immobilized protein, but while TINS uses a custom modified NMR spectrometer, WAC can get by with a much less pricey HPLC (though a mass-spectrometer seems nearly indispensible). It will be fun to see how WAC develops, and in particular whether it can be used to discover novel fragments against more challenging targets.
10 March 2011
Growing into closed pockets
Fragment-growing is a popular way to increase the activity of fragments, all the more so when there is an obvious place towards which to grow. In a paper published online in the J. Am. Chem. Soc., Iwan de Esch and colleagues at VU University Amsterdam describe the structural and thermodynamic consequences of one such effort, and conclude that binding in a certain normally closed pocket is enthalpically rather than entropically driven.
The researchers were interested in acetylcholine-binding protein (AChBP), a soluble and crystallizable homolog of an important class of ligand-gated ion channels. This is the same protein (and the same group) highlighted last year in the context of ligand efficiency hot spots. In the current work, a fairly potent fragment, compound 1, was co-crystallized with AChBP and the structure solved. This fragment binds in roughly the same position as the more potent natural product lobeline (compound 2), but lobeline contains a hydroxyphenethyl group that the fragment lacks (see figure). This moiety binds in a hydrophobic pocket that does not appear in the fragment complex due to the movement of a tyrosine residue. Recognizing the potential for the pocket to form, the researchers introduced this moiety into their own molecule, producing compound 3.

Gratifyingly, compound 3 binds about 50-fold more tightly than the initial fragment. This molecule was also co-crystallized with AChBP, and, as designed, the phenyl group binds in the “lobeline pocket". Moreover, compound 3 does not show a corresponding increase in affinity towards a version of AChBP from a different organism that does not have this pocket.
To correlate binding mode with thermodynamics, the researchers also characterized the binding of their compounds using surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC). ITC measures enthalpy directly, and performing SPR analyses at different temperatures can also be used to dissect enthalpic and entropic terms. The two methods generally concurred, though the numbers did jump around a bit, and in a few cases SPR predicted negative (ie, unfavorable) entropy where ITC suggested positive (favorable) entropy for the same compound.
Both SPR and ITC indicated that the increase in potency for compound 3 over compound 1 is driven by enthalpy, not entropy. This is somewhat unexpected, as the contacts made by compound 3 are largely hydrophobic, and the simplistic view is that such contacts are usually entropy-dominated. (The added hydroxyl in compound 3 doesn’t appear to be doing anything useful, and in fact removing it increases potency roughly three-fold). The researchers suggest that, for poorly solvated hidden pockets such as this, enthalpy may dominate. Perhaps also the protein rearrangement necessary to open the pocket is entropically costly.
There is much more data in the paper than can be summarized here, and the notion that ligands that induce conformational changes in proteins could be enthalpic rather than entropic binders is an intriguing hypothesis. However, as a vigorous debate last year demonstrates, it is still unclear whether knowing the answer – as scientifically interesting as it may be – will have practical implications for drug discovery.
The researchers were interested in acetylcholine-binding protein (AChBP), a soluble and crystallizable homolog of an important class of ligand-gated ion channels. This is the same protein (and the same group) highlighted last year in the context of ligand efficiency hot spots. In the current work, a fairly potent fragment, compound 1, was co-crystallized with AChBP and the structure solved. This fragment binds in roughly the same position as the more potent natural product lobeline (compound 2), but lobeline contains a hydroxyphenethyl group that the fragment lacks (see figure). This moiety binds in a hydrophobic pocket that does not appear in the fragment complex due to the movement of a tyrosine residue. Recognizing the potential for the pocket to form, the researchers introduced this moiety into their own molecule, producing compound 3.

Gratifyingly, compound 3 binds about 50-fold more tightly than the initial fragment. This molecule was also co-crystallized with AChBP, and, as designed, the phenyl group binds in the “lobeline pocket". Moreover, compound 3 does not show a corresponding increase in affinity towards a version of AChBP from a different organism that does not have this pocket.
To correlate binding mode with thermodynamics, the researchers also characterized the binding of their compounds using surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC). ITC measures enthalpy directly, and performing SPR analyses at different temperatures can also be used to dissect enthalpic and entropic terms. The two methods generally concurred, though the numbers did jump around a bit, and in a few cases SPR predicted negative (ie, unfavorable) entropy where ITC suggested positive (favorable) entropy for the same compound.
Both SPR and ITC indicated that the increase in potency for compound 3 over compound 1 is driven by enthalpy, not entropy. This is somewhat unexpected, as the contacts made by compound 3 are largely hydrophobic, and the simplistic view is that such contacts are usually entropy-dominated. (The added hydroxyl in compound 3 doesn’t appear to be doing anything useful, and in fact removing it increases potency roughly three-fold). The researchers suggest that, for poorly solvated hidden pockets such as this, enthalpy may dominate. Perhaps also the protein rearrangement necessary to open the pocket is entropically costly.
There is much more data in the paper than can be summarized here, and the notion that ligands that induce conformational changes in proteins could be enthalpic rather than entropic binders is an intriguing hypothesis. However, as a vigorous debate last year demonstrates, it is still unclear whether knowing the answer – as scientifically interesting as it may be – will have practical implications for drug discovery.
01 March 2011
Wedding announcement: Daiichi Sankyo and Plexxikon
Practical Fragments has highlighted several mergers and acquisitions of fragment-based companies over the years, but yesterday’s announcement that Daiichi Sankyo plans to acquire Plexxikon for $805 million in up-front cash plus an additional $130 million in near-term milestones is definitely the largest. You may recall that PLX-4032, Plexxikon’s B-Raf kinase inhibitor, is currently in Phase 3 trials and a favorite to be the first fragment-based drug approved.
According to FierceBiotech, Plexxikon has raised a total of $67 million in venture funding, so a number of investors are likely popping Champagne. This is a nice validation of the value of fragment-based drug discovery, so let’s all raise a glass!
According to FierceBiotech, Plexxikon has raised a total of $67 million in venture funding, so a number of investors are likely popping Champagne. This is a nice validation of the value of fragment-based drug discovery, so let’s all raise a glass!
Subscribe to:
Posts (Atom)

